Selection of Single Chain Variable Fragments
Specific for HBV DNA Polymerase
Selection of Single Chain Variable Fragments
Specific for HBV DNA Polymerase
Selection of Single Chain Variable Fragments
Specific for HBV DNA Polymerase
Using a Ribosome Display
by
Myung-Shin Lee
A Dissertation Submitted to The Graduate School of Ajou University in Partial Fullfilment of the Requirements for the Degree of
DOCTOR OF PHILOSOPHY
Supervised by Ho-Joon Shin, Ph.D.
Department of Medical Sciences The Graduate School, Ajou University
- ABSTRACT -
Selection of Single Chain Variable Fragments Specific for HBV
DNA Polymerase Using a Ribosome Display
Purposes: The purposes of this study are establishment of antibody selection system
by modifying previous ly reported ribosome display using eukaryotic translation system and selection of single chain variable fragments (scFvs) specific for the terminal protein (TP) of hepatitis B virus (HBV) DNA polymerase from a mouse scFv library by the established ribosome display.
Materials and methods: To establish an antibody selection system by ribosome
display, anti- DNA antibody was used. Anti-DNA antibody, 3D8 scFv, was synthesized in vitro by an in vitro transcription-translation system. The translated 3D8 scFv and the encoding 3D8 mRNA is connected to the ribosome and result in antibody-ribosome- mRNA complexes. The scFv-ribosome- mRNA complexes were selected by binding to their specific antigens. The eluted mRNAs from the complexes are reverse transcribed and re-amplified by PCR. To apply this system to select antibody from library, mouse was immunized with synthesized TP-peptide from HBV DNA pol TP domain to prepare antibody library. This TP-peptide encompasses the 57-80 amino acid residues of TP domain. The
antibody-ribosome-mRNA complex was synthesized by in vitro transcription/translation. These antibody-ribosome- mRNA complexes were panned four times aga inst TP-peptide. The enrichment of antibody to TP-peptide from the library was determined by radioimmunoassay. The fourth selected antibody genes from the library were cloned into expression vector and TP-specific clones were screened. The sequences of the selected genes were analyzed and binding activity was analyzed by ELISA.
Results: Anti-DNA antibody 3D8 selected specifically against ssDNA was used as a
model system. The selected 3D8 RNAs sequences from translation complexes were recovered by RT-PCR. By applying this model system, I enriched TP-peptide-specific scFv pools through four cycles of panning from immunized antibody library. The selected scFvs showed binding activity for both TP-peptide and HBV DNA polymerase protein.
Conclusions: Ribosome display using eukaryotic translation system for antibody
selection was established. Using this system, Anti- TP scFvs were successfully selected.
Keywords: Antibody, Hepatitis B virus, HBV DNA polymerase, Library, Ribosome display, scFv, Terminal protein (TP)
TABLE OF CONTENTS
TITLE PAGE --- 1 ABSTRACT--- 2 TABLE OF CONTENTS--- 4 LIST OF FIGURES--- 7 LIST OF TABLES--- 9 ABBREVIATIONS--- 10 I. INTRODUCTION--- 12 A. Hepatitis B virus--- 12B. HBV life cycle and replication--- 13
C. HBV DNA polymerase as the target for inhibition of replication--- 18
D. Antibody display methods--- 19
E. In vitro display--- 20
F. Ribosome display--- 21
G. Purposes and summary of study--- 27
II. MATERIALS & METHODS--- 29
A. Model system for ribosome display--- 29
1. Construction of VH/κ antibody fragment used for the control reaction of ribosome display--- 29
2. In vitro transcription of 3D8 VH/ê--- 34
3. In vitro translation of 3D8 VH/ê --- 34
4. ELISA using translated 3D8 VH/ê --- 35
5. Ribosome display using 3D8 VH/ê --- 36
5.1. Affinity selection--- 36
5.2. Reverse transcriptase - polymerase chain reaction (RT-PCR) --- 37
B. TP-specific scFv selection by using ribosome display--- 39
1. Immunization of Mice--- 39
2. Construction of VH/ê chain library and mRNA preparation--- 39
3. In vitro transcription--- 43
4. In vitro translation--- 43
5. Affinity selection--- 43
6. RT-PCR--- 44
7. Radioimmunoassay (RIA)--- 44
8. Cloning and expression--- 45
9. ELISA--- 46
10. Western blot analysis of scFv expression--- 47
11. Sequence analysis --- 47
III. RESULTS--- 48
A. Model system for ribosome display--- 48
1. In vitro translation --- 48
3. Ribosome display with 3D8 VH/ê --- 51
B. TP-specific scFv selection by using ribosome display--- 53
1. Immunization of mice--- 53
2. PCR amplification and preparation of mRNA--- 53
3. In vitro transcription and translation--- 56
4. Selection of TP-specific scFv gene --- 58
5. Soluble scFv production and sequence analysis--- 62
IV. DISCUSSION--- 68
V. CONCLUSIONS--- 73
BIBLIOGRAPHY--- 74
LIST OF FIGURES
Fig. 1. Hepatitis B virus life cycle--- 13
Fig. 2. HBV DNA in viral replication cycle--- 15
Fig. 3. Viral DNA synthesis in Hepadnavirus DNA replication--- 17
Fig. 4. The structure of antibody-ribosome- mRNA complex (ARM) complex--- 25
Fig. 5. Ribosome display cycle for antibody selection--- 26
Fig. 6. DNA structure used for ribosome display--- 32
Fig. 7. In vitro translated 3D8 VH/ê--- 50
Fig. 8. Binding activity of translated 3D8 VH/ê and ribosome display with 3D8 VH/ê--- 52
Fig. 9. Titration of immunized sera by indirect ELISA--- 54
Fig. 10. Amplified DNA fragments of VH DNA, kappa chain DNA and assembled VH/ê DNA in agarose gel electrophoresis--- 55
Fig. 11. In vitro translated library proteins detected by autoradiography--- 57
Fig. 12. Selection and amplification of VH/ê gene against TP-peptide --- 59
Fig. 13. Effect of internal primer in RT-PCR --- 60
Fig. 14. Analysis of the enrichment of TP-peptide-specific scFv pools--- 61
Fig. 15. Isolation of scFvs after 4th selection--- 64
Fig. 16. Binding activity of selected anti- TP scFvs--- 65
Fig. 18. Alignment of the amino acid sequence of selected TP-specific scFvs--- 67 Fig. 19. Direct comparison of immunoreactivity of selected anti-TP scFvs
by ribosome display and phage display--- 72
LIST OF TABLES
Table 1. Primers Used for 3D8C VH/κ construct--- 33 Table 2. Primers for VH/ê library and RT-PCR --- 42
ABBREVIATIONS
4-CN 4-Chloro-1-naphthol
ABTS 2,2'-Azino-bis(3-ethylbenzthiazoline-6-sulfonic acid) ARM complex Antibody-ribosome-mRNA complex
BCIP/NBT Bromo-chloro-indolyl-phosphate/nitroblue tetrazolium chloride
BSA Bovine serum albumin
cccDNA Covalently closed circular DNA
DEPC Diethyl pyrocarbonate
ELISA Enzyme-linked immunosorbent assay
ER Endoplasmic reticulum
HBV Hepatitis B virus
HCC Hepatocellular carcinoma
HRP Horseradish Peroxidase
IPTG Isopropyl-b-D-thiogalactopyranoside
PCR Polymerase chain reaction
pgRNA Pregenomic RNA
rcDNA Relaxed-circular DNA
RIA Radioimmunoassay
RT Reverse transcriptase
SDS Sodium dodecyl sulfate
ssDNA Single stranded DNA
I. INTRODUCTION
A. Hepatitis B virus (HBV)
Of the many viral causes of human hepatitis, few are of greater global importance than HBV. More than 2 billion people have been exposed to the HBV worldwide and estimated 50 million new cases are being diagnosed annually and 5-10% of adults and up to 90% of infants are becoming chronically infected. Healthy carriers still have the risk of developing chronic liver disease, including hepatocellular carcinoma (HCC). HBV is the leading cause of chronic hepatitis and up to 25% of these patients will eventually die of liver cirrhosis and its complications, including HCC. Worldwide, two million deaths a year are attributed to HBV-related liver disease and HCC.10, 23, 37, 56 HBV causes 80% of all liver cancer of human beings and is the second most important carcinogen, after smoking tobacco. There is an approximate 90% risk of becoming a persistent carrier following perinatal infection in infants born from HBe antigen positive carrier mothers, and a lower overall 30% risk in pre-school children. Only 5–10% of adults become persistent carriers following infection.22 Chemotherapy, such as alpha interferon or lamivudine remains the only treatment option for controlling chronic HBV infection once acquired, but none of the many different chemotherapeutic strategies used has proven consistently successful.34, 45, 49
B. HBV life cycle and replication
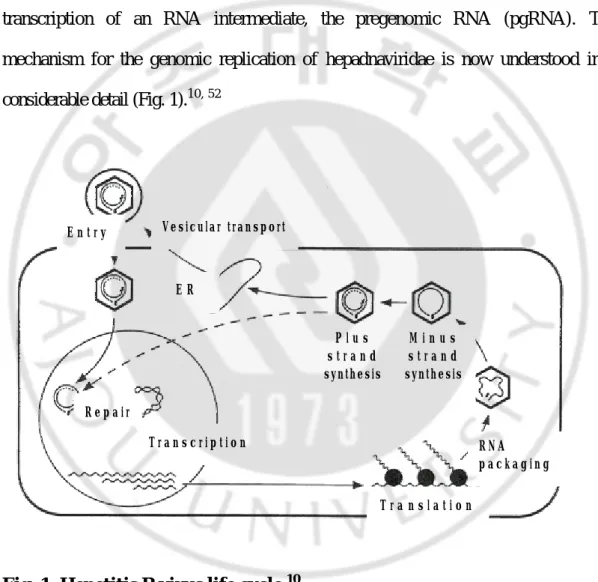
The life cycle of hepadnaviruses is characterized by the synthesis of a 3.2 kb partially double-stranded, relaxed-circular DNA (rcDNA) genome by reverse transcription of an RNA intermediate, the pregenomic RNA (pgRNA). The mechanism for the genomic replication of hepadnaviridae is now understood in a considerable detail (Fig. 1).10, 52
Fig. 1. Hepetitis B virus life cycle.10
V e s i c u l a r t r a n s p o r t E R P l u s s t r a n d synthesis E n t r y M i n u s s t r a n d synthesis R N A p a c k a g i n g T r a n s l a t i o n T r a n s c r i p t i o n R e p a i r
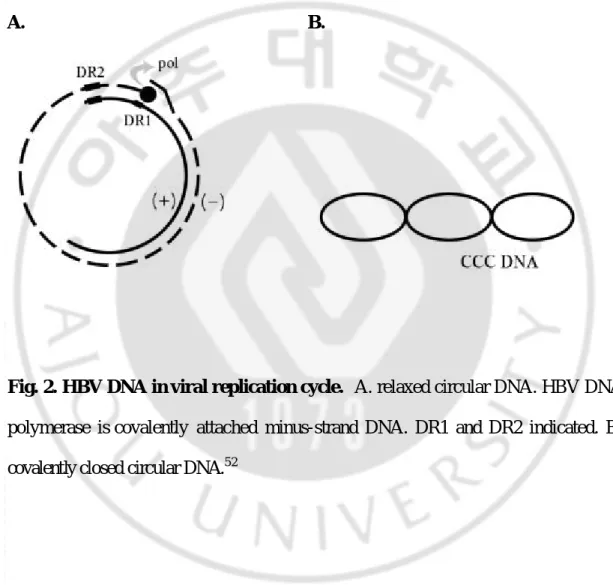
During initiation of infection, the viral rcDNA genome (Fig. 2A) with a HBV DNA polymerase attached to the 5' end of the minus-strand DNA and a short RNA attached to the 5' end of the plus-strand DNA, is converted into covalently closed circular DNA (cccDNA, Fig. 2B). The cccDNA serves as the template for transcription of viral mRNAs in nucleus. Resulting RNAs are translated to give rise to the polymerase, core, pre-surface/surface, and X gene products. One of these RNAs, the pgRNA that is capped, is polyadenylated, and has a large terminal redundancy, serves as the mRNA for the synthesis of core protein (nucleocapsid subunit) and the viral DNA polymerase. In cytoplasm, the viral DNA polymerase binds to the 5' end of its own viral pgRNAs, and the complex is then packaged into nucleocapsids, where viral DNA synthesis occurs. Within this structure, viral DNA synthesis is initiated; following minus-strand DNA synthesis, plus-strand DNA synthesis occurs. Once partially double-stranded DNA has been produced, progeny core bud into intracellular membranes such as the endoplasmic reticulum (ER) or proximal Golgi to acquire their glycoprotein envelops. These nucleocapsids can also migrate to the nucleus to increase the copy number of cccDNA. Enveloped virions are then secreted via the constitutive pathway of vesicular transport.
A. B.
Fig. 2. HBV DNA in viral replication cycle. A. relaxed circular DNA. HBV DNA
polymerase is covalently attached minus-strand DNA. DR1 and DR2 indicated. B. covalently closed circular DNA.52
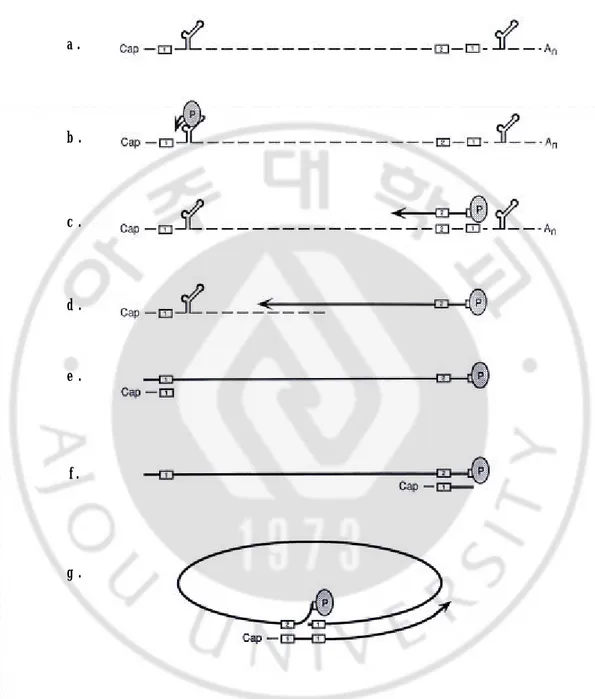
HBV DNA within the nucleocapsids is synthesized by following mechanism (Fig. 3). The HBV DNA polymerase binds to the epsilon stem- loop structure near the 5' end of pgRNA to facilitate packaging into nucleocapsids and initiation of reverse transcription by a protein-priming mechanism, utilizing a tyrosine located near the amino terminus of the reverse transcriptase itself (Fig. 3b). Following the synthesis of 4 bases, the polymerase translocates to the 3' end of the RNA template, where the 4 bases can anneal with complementary sequences (Fig. 3c). During the elongation of the minus-strand DNA to the 5' end of the pgRNA, pgRNA is degraded by the RNaseH activity of polymerase (Fig. 3d). When polymerase reaches the 5’ end of the template, its RNaseH activity leaves an RNA oligomer consisting of the 5' end 17 to 18 bases of the pregenome, including the CAP and DR1 (Fig. 3e). The remaining fragment then serves as the primer for plus-strand synthesis, usually following its translocation to DR2, with which it can hybridize because of the sequence identity between DR1 and DR2 (Fig. 3f). When the plus strand reaches the 5' end of the minus strand, a third translocation occurs to circularize the molecule and to allow continued plus-strand DNA elongation (Fig. 3g). This translocation may be facilitated by the short (~9 bases) terminal redundancy on the minus strand. The plus strand is not completed prior to virion release.
b . d . e . f . g . c . a .
Fig. 3. Viral DNA synthesis in Hepadnavirus DNA replication. See the text for details.10
HBV DNA polymerase as the target for inhibition of replication
HBV DNA polymerase has a central role in the life cycle of HBV and comprises four domains. From the amino terminus, the se are the terminal protein (TP), spacer, reverse transcriptase (RT), and RNaseH domains.10 As HBV DNA polymerase is poorly immunogenic in mice, the production of monoclonal antibodies that recognize this protein is currently limited. Some monoclonal antibodies that have been produced against human HBV DNA polymerase were raised against the TP, spacer and RNaseH domains. Of these antibodies, only TP domain-specific monoclonal antibodies were able to inhibit protein priming reaction.31, 60 These reports suggest that conserved structural features within the TP region of HBV DNA polymerase are important for its proper functioning. Within the TP domain of HBV DNA polymerase, a tyrosine residue is known to be the most important residue in protein priming and initiation of reverse transcription during viral replication.57 However, the previously reported monoclonal antibodies against the TP domain of human HBV polymerase recognize amino acids 8-20 and 20-30 within the TP domain.60 No antibody that recognizes the tyrosine 65 residue of HBV DNA polymerase where the protein-priming reaction occurs, has yet been identified. In order to produce monoclonal antibodies that recognize epitopes around the 65th residue of the TP domain of HBV DNA polymerase, I synthesized the TP-peptide that encompasses amino acid residues 57-80 of the TP domain of HBV DNA polymerase as a target antigen.
Antibody display methods
Antibodies or other proteins that have specific binding activity are useful tools for monitoring protein abundance and activity. During the past decade, several display strategies have been powerful and efficient tools for the selection and evolution of these kinds of proteins.4, 9, 36, 46 In these kinds of techniques, the connection of genotype and phenotype allows the enrichment of specific functional protein by using selection process, e.g. on immobilized target. In such selection systems, the target specific binding protein (phenotype) coupled to the specific sequence information (genotype) of members of libraries will be retained, while non-adherent proteins will be washed away. The gene encoding the selected protein can then be re-amplified for further evolution and analysis. The strategies that have been successfully developed are either cell-dependent, involving, for example, display on the surface of phage,58 bacteria11 or yeast,9, 27 or cell- free, as in the case of ribosome display17, 20 and mRNA display system39, 47 and so on.
The most commonly used technique is phage display in which each bacteriophage displays a unique peptide or protein on its surface to select ligands with high affinities in vitro.1, 24, 42 As would be expected, the affinity of ligands derived from this process increases with increasing complexity (or diversity) of the starting library.44, 55 The starting complexity of phage display lib raries is generally limited to <109 because of the bacterial transformation requirement. This limitation is also applied to other in vivo display systems. To overcome this limitation, various
techniques have been attempted.6, 29, 35, 55
In vitro display
In vitro methods employ well-defined, cell- free biochemical processes at all
steps. Potential advantages of these are larger libraries, fewer of the biases caused by cellular expression, more facile application of round-by-round mutagenesis technique. All of these means of applied evolution have specific advantages and limitations, and can be used in combination to exploit the synergy of the various approaches. The in
vitro methods can be categorized as 1) display format: relying on direct physical
linkage to associate genotype with phenotype, or 2) compartmentalization format: the co-sequestration of gene and gene product through the selection step. The display format has developed along two lines. The first relies on the continued association of mRNA with ribosomes and nascent protein, and has been called polysome display or ribosome display by different practitioners. This topic will be described later. Other display format was designed to join the mRNA and protein by a covalent bond, and does not require the continued presence of the ribosome to stabilize the complex. This method, which generally referred to as RNA display or RNA-peptide fusion, was introduced independently by two groups in 1997.39, 47 mRNA display takes advantage of the translation-terminating antibiotic puromycin, which functions by entering the A site of ribosomes and forming a covalent bond with the nascent peptide. By covalently attaching puromycin to the 3’ end of an mRNA, a covalent
link between a polypeptide and its encoding message can be achieved in situ during in vitro translation.32 These mRNA-peptide fusions can then be purified and subjected to in vitro selection. Some in vitro selection systems consisting of DNA-peptide fusion have been developed to stabilize the genotype. There are various type for these DNA-peptide fusions.28, 53 mRNA display procedure requires careful chemical synthesis and critical purification of puromycin-attached oligonucleotides, which must be ligated to the 3’ end of each mRNA in the sequence libraries. Failure to perform these manipulations appropriately leads to a reduction in the diversity of available libraries. Compartmentalized in vitro system was developed by Tawfik and Griffiths14, 54. This system used aqueous droplets in an oil/water emulsion mimicking cellular compartmentalization. This scheme maintains genotype-phenotype linkage and high concentrations of genes, RNA, proteins and substrates for more effective interaction of the components; and provides a much simpler, more defined micro-environment for controlling the selection conditions than does a living cell. Various compartmentalized in vitro systems were introduced7, 13 but these methods still required to be verified for practical use.
Ribosome display
Ribosome display is a one of the in vitro display methods. This method relies on non-covalent ternary complexes of mRNA, ribosome and protein.51 A fusion protein is constructed in which the domain of interest is fused to a C-terminal
tether, such that this domain can fold while the tether is still in the ribosome tunnel. This fusion construct lacks a stop codon at the mRNA level, thus preventing the release of mRNA and protein from the ribosome. High concentrations of magnesium and low temperature stabilize the ternary complex. In this system, relatively stable protein-ribosome- mRNA complexes, in which individual nascent proteins remain linked to their encoding mRNA, are formed by stalling ribosome at the end of translation. Protein- ribosome- mRNA linkage allows simultaneous selection of a desired protein and its encoding mRNA from a library (Fig. 3). The selected mRNA can then be converted into cDNA by reverse transcription reaction and amplified by polymerase chain reaction (PCR) (Fig. 4).
The size of a ribosome displa y library potentially very large, since the number of ribosome which can be concentrated into the given reaction volume is high. Thus, it should be possible to create very large libraries more quickly than for cell-dependent systems.
Ribosome display was first developed by Mattheakis et al. for the selection of peptides.36 To produce a population of stalled polysomes, agents such as rifampicin or chloramphenicol, which block prokaryotic translation, were used. A pool of DNA sequences encoding 1012 random decapeptides was applied for selection.12 Selection of the antibody fragment was developed by two groups, Hanes and Pluckthun17 and He and Taussig20. Hanes et al. int roduced additional features to prokaryotic system. One was the stalling of the ribosome through the absence of a stop codon. A number of additions were made to improve the yield of mRNA after
the polysome display cycle, including stem loop structures at the 5’ and 3’ ends of the mRNA, vanadyl ribonucleoside complexes as nuclease inhibitors, protein disulfide isomerase for folding of disulfide-bridged proteins, and an antisense nucleotide to inhibit ssrA RNA, which in the prokaryotic system causes release and degradation of proteins synthesized without stop codon. He et al. applied eukaryotic
in vitro expression to ribosome display. This methods derives from two experimental
results, namely the functional production of single chain antibodies in vitro in rabbit reticulocyte lysates40 and in the absence of a stop codon, individual nascent proteins remain associated with their corresponding mRNA as stable ternary polypeptide-ribosome-mRNA complexes.8, 21 According to the Hanes et al., the rabbit reticulocyte lysate system gave rise to lower amounts of functional complexes, lower enrichment factors.15 However, because the lysate are commercially available and contain a lower intrinsic RNase activity, the use of this system is easier and more convenient than using prokaryotic in vitro expression system. And it is also possible that different proteins might be expressed with different efficiencies in the two translation systems.2, 3, 41
In a model system using two distinct scFv fragments of an antibody, a 109 -fold enrichment of a specific scFv over the nonspecific scFv was achieved by five selection cycles of ribosome display, with an average enrichment of 100 per cycle.17 In a library selection, ribosome display was applied to the selection and simultaneous evolution of a scFv fragment binding with 40 pM affinity to a Gcn3p mutant peptide, using a library prepared from the spleen of immunized mice.16 And Starting from the
human combinatorial antibody library HuCAL, picomolar affinity binders to insulin were selected and evolved during ribosome display selection.18 All selected antibodies had accumulated many mutations during the PCR amplification cycles which is included in the ribosome display protocol, and the affinity of the antibodies had improved up to 40- fold compared to the antibodies initially present in the library. In a selection against an unusual DNA structure, namely the guanine quadruplex DNA, it was demo nstrated that antibodies with high specificity could be generated by ribosome display.50 Ribosome display selection was applied also to human antibody library using transgenic mice.19 Progesterone-bovine serum albumin was immunized to transgenic mice carrying human immunoglobulin loci in order to develop human antibody response. Human antibody fragment library from the immunized mice was prepared by recombination. The library was expressed in vitro and selected against progesterone-BSA. Selected antibody fragment have the affinity of ~10-8 M. The advantages of ribosome display are that, in comparison to phage display, larger libraries can be constructed without the transformation step, and the libraries can be further diversified by PCR during ribosome display.5 Ribosome display not only have applied for the selection for binding protein to a wide variety of target but also have great potential for the maturation of high-affinity protein binder and of protein stability.25, 26
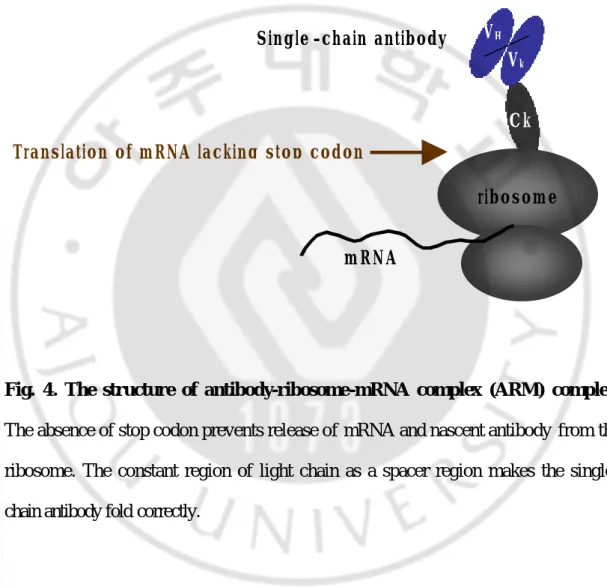
Fig. 4. The structure of antibody-ribosome-mRNA complex (ARM) complex.
The absence of stop codon prevents release of mRNA and nascent antibody from the ribosome. The constant region of light chain as a spacer region makes the single-chain antibody fold correctly.
ribosome
mRNA
Ck VH
Vk Single -chain antibody
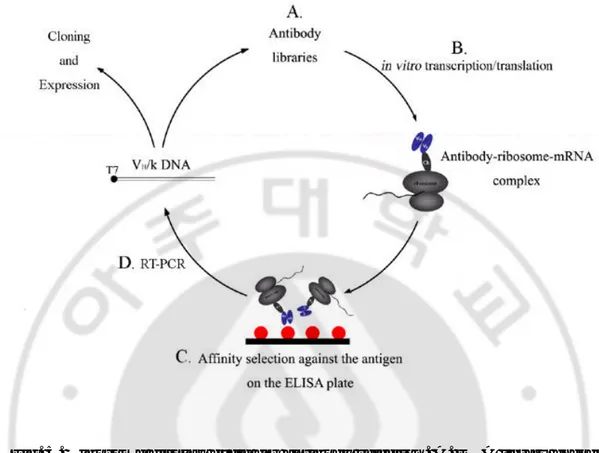
Fig. 5. Ribosome display cycle for antibody selection. A. DNA library encoding
the antibody fragments. It has genetically fused to a tether, which allows the protein to fold while the tether is still in the ribosomal tunnel. B. in vitro transcription and translation. The resulting construct, which lacks a stop codon, is transcribed in vitro into mRNA and further translated in vitro. A ribosomal pausing is induced during the translation. That result in stable ternary complexes of antibody-ribosome- mRNA (ARM complexes) formed. C. Affinity selection against the antigen on the ELISA plate. The ARM complexes are directly used to select by binding assay on the immobilized target. The mRNA of the bound complexes is rescued by dissociating the ribosome with EDTA. D. RT-PCR. A reverse transcription reaction followed by PCR yields the genetic information of the selected clones. These clones can then be analyzed or used as input for the next selection round.
Purpose and summary of this study
In this study, I established the ribosome display and then selected useful TP-specific scFv by established ribosome display.
Although ribosome display has many advantages theoretically, only 4 reports have been published about antibody selection from a library using ribosome display.16, 18, 19, 50 Moreover, only one of them exploited a eukaryotic translation system.19 To select anti- TP antibody using ribosome display, it is necessary to establish ribosome display and to test that antibody selection by ribosome display using eukaryotic translation system works properly. After establishing ribosome display, anti-DNA antibody, 3D8 scFv that has binding activity to single stranded DNA (ssDNA), anti- TP scFv was selected from immunized mouse library. Synthetic TP-peptide was used to immunize mice for library construction. mRNA library was obtained from the spleen of the immunized mouse. Recombinant antibody library DNA was constructed by RT-PCR and assembly PCR reaction. From this library DNA, the antibody-ribosome-mRNA (ARM) complexes were prepared by in vitro transcription and in vitro translation. ARM complexes were specifically selected against TP-peptide. After enrichment of antibody library to the TP-peptide by four repeated selection, library was inserted to expression vector. The selected scFvs had binding affinity, for not only the TP-peptide, but also for functional HBV DNA polymerase protein expressed from baculovirus-infected insect cells.
established and this system was successfully applied to select TP-specific antibody from an antibody library.
II. MATERIALS AND METHODS
A. Model system for ribosome display
1. Construction of VH/ê antibody fragments used for the control reaction of
ribosome display
As a model system for ribosome display, previously isolated ssDNA binding scFv (3D8) antibody fragment was used.30 In order for the scFv fragments to be fold outside of the putative ribosomal tunnel, the kappa chain constant region was used as a spacer as schematically shown in Fig. 6. For in vitro transcription and translation, the construct contained the T7 promoter and Kozac sequence. For optimal analysis, such as ELISA, polyhistidine (His6) affinity tag was connected to the 5’ end of the construct. The construct was prepared by assembly PCR. The primers for construction were listed in Table 1. First, 3D8 scFv DNA and the constant region were amplified separately. The 3D8 scFv gene was amplified from pIg20 3D8
plasmid by using 10 pmole primer of 5’ HIS3D8/back
(5’-GACCACCATGGACCATCATCATCATCATCATGAGGTCCAGCTGCAGCAG-3’) and 3’ 3D8/for (5’-GTTGGTGCAGCATCAGCCCGTTTTATTTCCAGCTTGGTC-3’) in a reaction volume of 50 ㎕ buffer [10 mM Tris-Hcl (pH 8.3), 40 mM potassium chloride, 10 mM DTT, 1.5 mM magnesium chloride] containing 2 U of
pfu DNA polymerase (Bioneer, Daejun, Korea). After 5 min of denaturation at 94 ,
samples were amplified for 25 cycles (1 min at 94℃, 1 min at 55℃ and 1 min at 72℃). For Ck DNA amplification, RNA was isolated from a mouse spleen. In brief, the spleen from BALB/c mouse was removed and the tissue was teased apart using either sterile forceps and then disrupt between microscope slides glass to produce a single cell suspension of lymphocytes and erythrocytes. Total RNA was extracted from the cell suspension by using RNA extraction kit (Amersham Biosciences, Piscataway, NJ, USA). First strand cDNAs were synthesized from prepared total RNA using superscript II RNaseH- reverse transcriptase (Invitrogen, Carlsbad, CA, USA). Ck DNA was amplified from the cDNA by using 10 pmole primer of 5’
Ck/back (5’-AAACGGGCTGATGCTGCA-3’) and 3’ Ck/for_XmaI
(5’-TCCCCCCGGGCTCTAGAACACTCATTCCTGTTGGAGCT-3’) in a reaction volume of 50 ㎕ buffer (10 mM Tris-Hcl (pH 8.3); 40 mM potassium chloride; 10 mM DTT; 1.5 mM magnesium chloride) containing 2.5 U of Taq DNA polymerase (Bioneer Co). After 5 min of denaturation at 94 , samples were amplified for 25 cycles (1 cycle is 30 sec at 94℃, 30 sec at 55℃ and 30 sec at 72℃). 3D8/for and Ck/back primers are designed in such a way that both the scFv DNA at its 3’ end and the spacer DNA at its 5’ end contain an identical sequence of 18 nucleotides. The PCR fragments were purified by QIAexII gel extraction kit (Qiagen, Stanford Valencia, CA, USA) and used for assembly PCR. The assembly PCR was conducted in a 50 ㎕ PCR mixture containing 5U of pfu DNA polymerase for 25 cycles (1 cycle is 1 min at 94℃, 1 min at 55℃ and 1 min at 72℃) with the primer ST7/back and
Ck/for_XmaI. The amplified product was purified by gel extraction and cloning to pUC18. Amplified DNA fragment was cloned into SmaI (New England Biolabs, Beverly, MA, USA) of pUC18 vector. In brief, the amplified DNA fragment was mixed with pUC18 vector in 10 ㎕ of 1x ligation buffer and then incubated at 16 for overnight in the presence of 1 U of T4 DNA ligase (USB, Cleveland, OH, USA). After the ligations mixture was transformed into E . coli DH5á. Five were picked and grown at 37℃ for overnight in 5 ㎖ of ampicillin containing LB broth. The plasmids were isolated using plasmid mini-prep kit (Bioneer) and sequenced with the ABI Perkin Elmer automated DNA sequencer (Applied Biosystems, Foster, CA, USA). Using the pUC sequencing primer (5’-GTTTTCCCAGTCACGAC-3’) and the pUC reverse sequencing primer (5’-AGCGGATAACAATTTCACACAGGA-3’). The plasmid with expected sequences was digested with SmaI. The restricted DNA was extracted and purified by a subsequent ethanol precipitation for in vitro transcription.
A.
B.
5’-GGTACCCGCAGCTAATACGACTCACTATA GGAACAGACCACCATGGAC T7 promoter Kozac sequence CATCATCATCATCATCATGAG……
H H H H H H
Polyhistidine (His6) tag 3D8 scFv
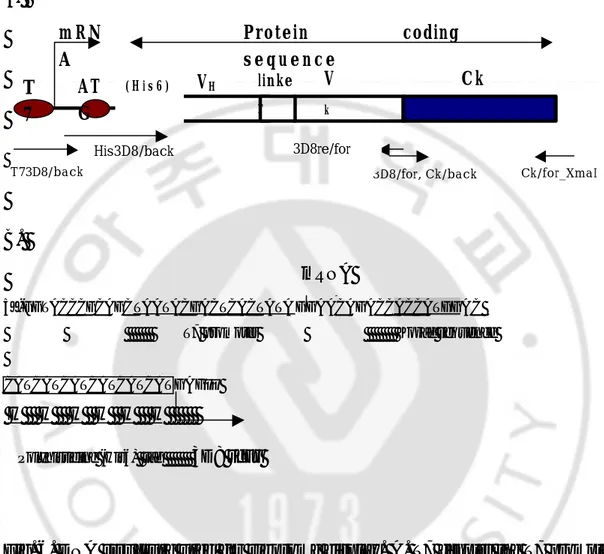
Fig. 6. DNA structure used for ribosome display. A. T7 denotes the T7 promoter,
ATG the protein initiation sequence, VH the variable region of heavy chain, Vk variable region of kappa chain, Ck the constant region of kappa chain that act as a spacer region connecting the folded protein to the ribosome. Large arrows indicate the transcriptional start and protein coding sequence. Small arrows are the primers used for this system. B. DNA sequence of promoter, transcription initiation site and translation initiation site used for ribosome display.
T 7 V k Ck m R N A Protein coding s e q u e n c e Ck/for_XmaI ( H i s 6 ) VH linke r 3D8/for, Ck/back T73D8/back His3D8/back AT G mRNA 3D8re/for
Table 1. Primers used for 3D8C VH/κ construct
A. Primers used to generate the 3D8 scFv DNA T73D8 /back
5’-GCAGCTAATACGACTCACTATAGGAACAGACCGACCACCATGGA CCATC-3’
The T7 promoter sequence is underlined. Kozac sequence is indicated as box.
Hi s3D8/back
5’-GACCACCATGGACCATCATCATCATCATCATGAGGTCCAGCTGCA GCAG-3’
3D8/for
5’-GTTGGTGCAGCATCAGCCCGTTTTATTTCCAGCTTGGTC-3’
B. Primers used to generate the complete mouse kappa light chain Ck/back
5’-AAACGGGCTGATGCTGCA-3’
Ck/for_XmaI
5’-TCCCCCCGGGCTCTAGAACACTCATTCCTGTTGGAGCT-3’
2. In vitro transcription of 3D8 VH/κ
3D8 VH/κ mRNA that encodes recombinant antibody that composed of 3D8 scFv and kappa chain constant region was obtained by in vitro transcription from the constructed structure. Briefly, approximately 500 ng of prepared DNA was added to the transcription mixture containing 10 ㎕ of 10 mM NTP mix (2.5 mM ATP, 2.5mM CTP, 2.5mM UTP, 1.25mM GTP; Invitrogen), 5 ㎕ of 10 mM RNA Capping analog, 2.5 ㎕ of 10 mM DTT, 40 U of RNase inhibitor (Invitrogen) and 30 U of T7 RNA polymerase (Bioneer). The mixture was incubated at 37℃ for 1 hr and the reaction was stopped by phenol/chloroform extraction. The transcripts were precipitated using 5 M ammonium acetate and dissolved in diethyl pyrocarbonate (DEPC)-treated water.
3. In vitro translation of 3D8 VH/κ
Flexi rabbit reticulocyte lysate (Promega, Madison, WI, USA) was used to in
vitro translation reaction. About 5 ㎍ of 3D8 VH/ê RNA was denatured at 65℃ for 10 min. The reaction mixture containing 1.6 ㎕ of 2.5 M potassium chloride, 1.4 ㎕ of 25 mM magnesium chloride, 1 ㎕ of 1 mM methionine or [35S] labeled methionine (Amersham Biosciences), 1 ㎕ of 1 mM amino acid mixture without methionine, 40 U of RNase inhibitor and 33 ㎕ of rabbit reticulocyte lysate was added to the prepared RNA, and incubated at 30℃ for 6 min. After in vitro translation, the
translated protein was detected by Western blot analysis using anti-His antibody or autoradiography for radiolabeled proteins. Five microliters of translated protein were suspended in 20 ㎕ of sample buffer (62.5 mM Tris pH 6.8; 10% glycerol; 10% 2-mercaptoethanol; 3% SDS and 0.1% bromophenol blue), boiled for 2 min prior to loading onto a gel. The prepared protein was subjected to SDS-PAGE on a 12% polyacrylamide gel. After SDS-PAGE, the gel was either transferred to a nitrocellulose membrane (Schleichr & Schuell, Keene, NH, USA) for Western blot or fixed and dried for autoradiography. For Western blot analysis, the transblotted membrane was blocked for 1 hr at 25℃ with blocking solution (3% (w/v) bovine serum albumin in PBS) and then incubated for 1 hr at 25℃ with anti-His antibody (1:1,000 dilution with blocking solution; Qiagen). After washing, alkaline phosphatase conjugated goat anti- mouse IgG antibody was treated (1:2,000 dilution with blocking solution; Pierce, Rockford, IL, USA) for 1 hr at 25℃. BCIP/NBT (bromo-chloro- indolyl-phosphate/nitroblue tetrazolium chloride; Sigma-Aldrich, Milwaukee, MI, USA) was used as AP substrate to visualize immunoreactivity. For autoradiography, the gel was fixed in 7% acetic acid for 10 min and rinsed briefly in deinonized water. The gel was dried onto a piece of Whatman 3MM paper using a gel dryer and exposed to a X-ray film at -70℃ for overnight prior to development.
4. ELISA using translated 3D8 VH/κ
by ELISA. Microtiter plates (Costar, High Wycombe, UK) were coated with 50 of l ssDNA solution (5 ㎍/㎖ in PBS) or PBS as negative control at 4℃ overnight. The coated plates were washed with PBS 3 times and blocked with 3 % (w/v) BSA in PBS for 1 h at 37℃. After washing with PBS, translated protein in ice-cold PBSM (PBS containing 5 mM magnesium chloride) was added and incubated for 1 hr at 4℃. The wells were washed with PBST 3 times and then anti-His antibody (1:1,000; Qiagen) as secondary antibody was added to each well. After incubation for 1hr at RT, the plate was washed 3 times with PBST. HRP-conjugated anti- mouse IgG antibody (Zymed Laboratories., South San Francisco, CA, USA) was reacted for 1h at RT. After washing as described above, 100 ㎕ of ABTS (2,2'-Azino-bis(3-ethylbenzthiazoline-6-sulfonic acid); Amersham Biosciences) substrate solution per well was added for measuring at A405 .
5. Ribosome display using 3D8 VH/κ
5.1. Affinity selection
Microtiter plates (Costar) were coated overnight at 4℃ with 50 ㎕ of ssDNA solution (5 ㎍/㎖ in PBS) or PBS as negative control. The coated plates were washed, blocked for 1 hr at 37℃, washed with PBSM 3 times, and incubated on ice for at least 10 min.
(Promega ) was used. The reaction mixture was prepared as described in Section 1.3. The library mRNA was incubated for 5 min at 65℃, and then added to the in vitro translation reaction mixture. After 20 min of in vitro translation at 30℃, ice-cold buffer (PBS containing 5 mM magnesium chloride, and 1.5% (w/v) BSA) was immediately added to the mixture. This mixture was transferred to the prepared plate that coated with ssDNA and washed with PBSM. The plate was incubated for 1 hr in a cold room on ice. After three washes with ice-cold PBSTM (PBS containing 5 mM magnesium chloride, and 0.05% (v/v) Tween 20) and two washes with ice-cold PBSM, the retained ribosomal complexes were dissociated with 200 ㎕ of EB20 buffer (PBS containing 20 mM EDTA) for 10 min on ice. The mRNA was isolated from the eluted solution by an RNA isolation kit (Roche Applied Science, Indianapolis, IN, USA) according to the manufacturer’s instruction.
5.2. Reverse transcriptase-polymerase chain reaction (RT-PCR)
Selected mRNA was reverse transcribed to cDNA using superscript II RNaseH- reverse transcriptase (Invitrogen). The elution buffer containing selected mRNA was denatured at 70℃ for 10 min and then added to final volume of 10 ㎕ of containing 2 ㎕ of 5× buffer, 1 ㎕ of 10 mM dNTP, 1 ㎕ of 10 mM DTT, 20 U of RNase inhibitor (Invitrogen), Ck/for primer (5’-GCTCTAGAACACTCATTCCTGT TGGAGCT-3’) and 0.5 ㎕ of superscript II RNaseH- reverse transcriptase. The
reaction mixture was incubated at 42℃ for 1 hr and 70℃ for 15 min. The VH and linker region of 3D8 scFv was amplified in a 50 ㎕ PCR mixture containing 5 ㎕ of 10× buffer, 2 ㎕ of RT reaction mixture, 10 mM dNTP, 5 pmol of His3D8/back primer (Table 1) and 5 pmol of 3D8re/for (5’-CAGCCAGGGAGGATGGAGAC-3’) primer and 5 U of Taq DNA polymerase (Genesis, Daejun, Korea). After 5 min of denaturation at 95℃ for 2 min, sample was amplified for 30 cycles (1 cycle is 20 sec s at 95 , 40 sec at 55 , 1 min at 68 ). The reaction products were analyzed by electrophoresis in a 1% agarose gel.
B. TP-specific scFv selection by using ribosome display
1. Immunization of Mice
Biotinylated TP-peptide was prepared by chemical synthesis and has the sequence biotin-KVGNFTGLYSSTVPIFNPEWQTPS. Neutravidin (Sigma-Aldrich) was conjugated to TP-peptide as a carrier protein by the N terminally linked biotin. Female BALB/c mice were given an intraperitoneal injection of conjugated protein (80 ㎍ of TP-peptide and 400 ㎍ of neutravidin), emulsified in the same amount of complete Freund’s adjuvant (Sigma-Aldrich), followed by one intraperitoneal injection with incomplete Freund’s adjuvant (Sigma-Aldrich) over a 3-week period. The mice were boostered with an intraperitoneal injection twice at 2-week intervals. Total 4 mice were immunized. The sera were collected for ELISA from their tails before euthanasia. A spleen from an immunized mouse that had the highest titer of sera was removed and cells were isolated.
2. Construction of VH/ê chain library and mRNA preparation
Splenic cells were prepared by the spleen from the immunized BALB/c mouse was removed and teasing apart using a sterile forceps, and squeezing between microscope slides. From these cells total RNA was extracted from this cell suspension using RNA extraction kit (Amersham Biosciences). The mRNA was
prepared from extracted total RNA using an oligo(dT)-cellulose column. DNAs encoding the mouse VH and VL chains were obtained as follows. The reaction mixture containing 10 ㎕ of 5× reaction buffer, 1 ㎕ of 10 mM dNTP mix, 2 ㎕ of 25mM magnesium sulfate, 10 pmol of 5’ primers T7Ab/back (5’- CAGCTAATACGACTCACTATAGGAACAGACCACCATG(GC)AGGT(GC)CA(G C)CTCGAG(GC)AGTCTGG-3’), 10 pmol of 3’ primer VH/for (5’- TGAGGAGACGGTGACCGTGGTCCCTTGGCCCC-3’), 5 U of AMV reverse transcriptase and 5 U of Tfl DNA polymerase were used for amplification of DNA encoding VH. The reaction mixture was incubated at 48℃ for 45 min and then amplified 35 cycle of PCR reaction (1 cycle is 30 sec at 94℃, 1 min at 54℃, and 2 min at 68℃). Primers of Vk2/back (5’-GACATTGAGCTCACCCAGTCTCCA-3’) and Ck/for (5’-GCTCTAGAACACTCATTCCTGTTGGAGCT-3’) were used for amplification of DNA encoding the kappa chain of a mouse antibody. A 93-bp DNA linker (Amersham Biosciences), encoding (Gly4Ser)3, was amplified with primers LINKBACK GGGACCACGGTCACCGTCTCCTCA-3’) and LINKFOR (5’-TGGAGACTGGGTGAGCTCAATGTC-3’) using Taq polymerase (Genesis) with 25 cycles of PCR (1 cycle is 1 min at 94℃, 1 min at 55℃, and 1 min at 72℃). After gel purification with a QIAexII gel extraction kit (Qiagen), 15 ng of VH DNA, 20 ng of linker DNA, and 50 ng of kappa chain DNA were mixed with 25 ㎕ of PCR mixture containing 2.5 ㎕ of 10x buffer, 0.5 ㎕ of 10 mM dNTP mix and 5 U of Taq polymerase (Genesis). For joining the linker DNA with VH and k-chain DNA, reaction mixture was cycled 25 times (1 cycle is 1 min at 94℃, 2 min at 60℃, 2 min
at 72℃). For amplification of assembled DNA, 5 µl previous reaction product was subjected to PCR in a total volume of 50 ㎕ containing 1× reaction buffer, 0.2 mM dNTP, 10 pmol of 5’ primer T7/back, 10 pmol of 3’ primer Ck/for and 5 U Taq polymerase. The sample was amplified for 25 cycles in a DNA thermal cycler (Applied Biosystems, Foster, CA) with the following cycle profile: 1 min at 94℃, 1 min at 55℃ and 1 min at 72℃. Finally an extension step of 5 min at 72℃ was performed. After analyzing on a 1% agarose gel, the amplified DNA was purified by phenol/chloroform extraction and precipitated using 3 M sodium acetate (pH 6.8) and dissolved in sterile water.
Table 2. Primers for VH/κ library and RT-PCR
A. Primers used to generate the VH/κ library
T7/back 5’-CAGCTAATACGACTCACTATAGGAACAGACCACCATG(GC)AGGT (GC)CA(GC)CTCGAG(GC)AGTCTGG-3’ VH/for 5’-TGAGGAGACGGTGACCGTGGTCCCTTGGCCCC -3’ Vk2/back 5’-GACATTGAGCTCACCCAGTCTCCA-3’ Ck/for 5’-GCTCTAGAACACTCATTCCTGTTGGAGCT-3’ LINKBACK 5’-GGGACCACGGTCACCGTCTCCTCA-3’ LINKFOR 5’-TGGAGACTGGGTGAGCTCAATGTC-3’ B. Primers RT-PCR D2 5’-CGTGAGGGTGCTGCTCAT-3’ D3 5’-GCCATTTTGTCGTTCACTGCCATC-3’ D4 5’-CTGGATGGTGGGAAGATGG-3’
3. In vitro transcription
About 500 ng of the prepared DNA was used to the 50 ㎕ of transcription reaction. In vitro transcription was conducted same as described protocol 1.2. in vitro transcription of 3D8 VH/κ.
4. In vitro translation
In vitro translation and autoradiography of antibody library was conducted same as described protocol 1.3. in vitro translation of 3D8 VH/κ.
5. Affinity selection
Microtiter plates (Costar) were coated at 4 overnight with 50 ㎕ of TP-peptide solution (1 ìM in PBS) or PBS. The coated plates were washed with PBS and blocked with sterilized 10 % (w/v) skim milk in PBSM (PBS with 5 mM Magnesium chloride) for 30 min at 25℃. After washing with PBS, it was blocked again with blocking buffer (5% (w/v) BSA in PBSM) for 2 hr, followed by washing with PBSM 3 times and incubation on ice for at least 10 min.
Fifty microliter of in vitro translated products were immediately mixed with 150 ㎕ of ice-cold buffer (PBS containing 5 mM magnesium chloride, and 1.5% (w/v) BSA), mixture was added to the PBS-coated microtiter plate, and incubated at
4℃ for pre-binding. After pre-binding, the supernatant was transferred to a TP-peptide coated well. The plate was incubated for 1 hr on ice, washed 3 times with ice-cold PBSTM (PBS containing 5 mM magnesium chloride, and 0.05% (v/v) Tween 20) and twice with ice-cold PBSM, The retained ribosomal complexes were dissociated with 200 ㎕ of EB20 buffer (PBS containing 20 mM EDTA) for 10 min on ice. The mRNA was isolated from the eluted solution using an RNA isolation kit (Roche Applied Science, Indianapolis, IN, USA), as described by the manufacturer.
6. RT-PCR
RT-PCR was conducted same as protocol 1.5.2. RT-PCR except primers. T7/back (5’- CAGCTAATACGACTCACTATAGGAACAGACCACCATG(GC)AG GT(GC)CA(GC)CTCGAG(GC)AGTCTGG-3’) and Ck/for (5’-GCTCTAGAACA CTCATTCCTGTTGGAGCT-3’) primers used in reaction. Another downstream primers was used, D2 (5’- CGTGAGGGTGCTGCTCAT-3’) primer in the second cycle, D3 (5’-GCCATTTTGTCGTTCACTGCCATC-3’) in the third cycle, and D4 (5’-CTGGATGGTGGGAAGATGG-3’) in the fourth cycle for nested PCR.
7. Radioimmunoassay (RIA)
Microtiter plates (Costar) were prepared as described for affinity selection. The translation mixture was prepared as described for affinity selection, except 1 ㎕
of methionine was substituted by 2 ㎕ [35S] methionine (50 ìCi/ml). After translation, 150 ㎕ of ice-cold buffer (PBS containing 5 mM magnesium chloride, and 1.5 % (w/v) BSA) was added to the translation mixture. Binding was performed for 1 hr at 4 . After washing five times with PBST, bound protein was eluted with 200 ㎕ of 4% (w/v) sodium dodecyl sulfate (SDS) at room temperature for 10 min. Bound proteins were quantified with a Microbeta TriLux scintillation counter (PerkinElmer Life and Analytical Sciences, Boston, MA, USA).
8. Cloning and expression
To amplify the scFv DNA sequences from selected DNA, a forward primer,
VH/back_SfiI (5’
GTCGTCGCAACTGCGGCCCAGCCGGCCATGGCC(GC)AGGT
(GC)CA(GC)CTCGAG(GC)AGTCTGG 3’) and a reverse primer, Vk/For_NotI (GAGTCATTCTGCGGCCGCTGCAGCATCAGCCCGTTT) were used in PCR (restriction sites SfiI and NotI, respectively, are underlined). The PCR reaction was performed by pfu DNA polymerase (Bioneer Co) for 25 cycles (94℃ for 30 s, 55℃ for 40 s, 72℃ for 1 min). The amplified scFv DNA and pCANTAB5E vector (Amersham Biosciences) were digested with SfiI (New England Biolabs) and NotI (New England Biolabs) and purified using the QIAexII gel extraction kit (Qiagen). Ligations of prepared insert DNA and pCANTAB5E vector were carried out using T4 DNA ligase. The insert DNA was mixed with pCANTAB5E vector in 10 ㎕ of
1× ligation buffer and then incubated at 16℃ for overnight in the presence of 1 U of T4 DNA ligase (USB, Cleveland, OH, USA). The ligations were transformed into E.
coli TG1 and soluble proteins were expressed from each clone. Briefly, each single
colony was cultured in 5 ㎖ of 2×-YT medium with 100 ㎍/㎖ ampicillin and 0.1% (w/v) glucose at 30℃ with 250 rpm shaking until they reached an absorbance of 0.7 at 600 nm. Isopropyl-b-D-thiogalactopyranoside (IPTG) was added to obtain a final concentration of 1 mM , and the cells were incubated at 30℃ overnight with shaking at 130 rpm. Cells were pelleted and resuspended in 0.5 ml ice-cold 1× TES buffer (0.2 M Tris-HCl (pH 8.0), 0.5 mM EDTA, 0.5 M sucrose) and 0.75 ㎖ ice-cold 1/4× TES buffer. After incubation on ice for 30 min, the cells were pelleted by centrifugation at 10,000 rpm for 10 min and the supernatant retained as periplasmic extracts containing the soluble scFvs.
9. Enzyme-l inked i mmunosorbent assay (ELISA)
To screen TP-specific scFvs, periplasmic extracts from each clone were analyzed by ELISA. Microtiter plates (Costar) were coated with synthetic TP-peptide or human HBV DNA polymerase expressed in a baculovirus- infected insect system at 37℃ for 2 hr and 5% (w/v) BSA was used for blocking. After washing, periplasmic extracts with a final concentration of 1% (w/v) BSA were added to the well and incubated overnight at 4℃. To determine the amount of soluble scFv antibody bound, the microtiter plate was incubated with 100 ㎕ of mouse anti- E tag
HRP conjugate (Amersham Biosciences) in blocking buffer (1:4,000) at room temperature for 1 hr. After washing, ABTS (Sigma-Aldrich) was used as a substrate, and absorbance was determined using a microtiter plate reader with a 405 nm measurement filter.
10. Western blot analysis of scFv expression
Periplasmic extracts from selected anti- TP produc ing clones were subjected to SDS-PAGE on a 12% polyacrylamide gel. Prestained SDS-PAGE standards (New England Biolabs) were used to calibrate protein mobilities. After SDS-PAGE, the proteins were transferred to a nitrocellulose membrane (Schleichr & Schuell). The transblotted membrane was blocked for 1 hr at RT with blocking solution (2% (w/v) skim milk in PBS) and then incubated for 1 hr at RT with peroxidase-conjugated mouse anti- E tag (1:1,000 dilution with blocking solution). 4-CN (4-Chloro-1-naphthol, Sigma-Aldrich) was used as a peroxidase substrate to visualize immunoreactivity.
11. Sequence analysis
Plasmid DNA from anti-TP producing clones was isolated from E. coli TG1. The scFv DNA was sequenced on both strands with the pCANTAB5E sequence primer set (Amersham Biosciences) using an ABI Perkin Elmer 373A automated
III. RESULTS
A. Model system for ribosome display
1. 3D8 VH/ê sequence and in vitro synthesized 3D8 VH/ê
After recombinant PCR and cloning, nucleotide sequencing was performed to check frameshift mutation or stop codon. Assembled DNA was in vitro transcribed by T7 RNA polymerase, and then it was treated with DNaseI to remove the template DNA. The synthesized RNA was checked by agarose gel electrophoresis (Fig. 7A).
To obtain the 3D8 VH/κ antibody fragment, the synthesized RNA was in
vitro translated by rabbit reticulocyte lysate. Two strategies were used to evaluate to
assess if 3D8 VH/κ was synthesized. The translated 3D8 VH/κ protein was labeled with [35S] methionine, and subjected to SDS-PAGE followed by autoradiography (Fig. 7B). The 3D8 VH/κ mRNA translation product migrated as an intact band with molecular weight of 40 kDa approximately. The luciferase RNA translation product was approximately 65 kDa as a positive control. Translation mixture without mRNA or containing cycloheximide which did not show any radioactive band was employed as a negative control. The molecular weight of the synthesized 3D8 VH/κ was similar to those theoretically calculated (40 kDa).
at 5’ end, the 3D8 VH/κ protein could be detected by Western blot with anti- His antibody. A band of approximate 40 kDa was detected by Western blot from the translation mixture with 3D8 VH/κ RNA (Fig. 7c). Another strong band about 45 kDa molecular weight was detected but also detected in negative control reaction, such as translation mixture without RNA or with luciferase RNA. This band could be a protein in rabbit reticulocyte lysate that is reacted with anti-His antibody.
1kb 500 bp
A. B.
Fig. 7. The synthesized 3D8 VH/ê. A. 3D8 VH/ê RNA. After RNA transcription, DNA was removed by DNaseI treatment. The transcribed RNA was checked by gel electrophoresis. B. In vitro translated 3D8 VH/ê. After translation reaction, the translation mixture was subjected to the 12% SDS-PAGE, and detected by autoradiography. The translated 3D8 VH/ê protein was indicated by an arrow. [14C] protein molecular weight marker (lane M), luciferase as control (lane 1) and 3D8 VH/ê (lane 2) are shown. C. Western blot of 3D8 VH/ê. Translation of 3D8 VH/ê RNA (lane 1), translation without RNA (lane 2), and translation of 3D8 VH/ê RNA with cycloheximide (lane 3) are shown.
220K 97K 66K 45K 30K 20K C.
2. Binding activity of translated 3D8 VH/ê
The binding activity of translated 3D8 VH/κ protein was analyzed by ELISA (Fig. 8A). The translation mixture with 3D8 VH/κ RNA was specifically bound to ssDNA but translation mixture without RNA or with cycloheximide which inhibit translation did not show any reactivity to ssDNA.
3. Ribosome display with 3D8 VH/κ
After it was confirmed that in vitro translated 3D8 was bound to ssDNA specifically, it was checked whether 3D8 gene was specifically selected by ribosome display. The 3D8 gene could be selected from ssDNA immobilized surface. After selection with ssDNA coated surface, eluted RNA was amplified by reverse transcription PCR and analyzed by agarose gel electrophoresis. ssDNA reacting translation mixture with 3D8 VH/κ RNA showed a clear amplified band (Fig. 8B). A faint band was observed when translation mixture with 3D8 VH/κ RNA was reacted with bovine serum albumin and no band was detected from translation mixture without 3D8 VH/κ RNA or with cycloheximide. The binding activity determined by ELISA was consistent with the result of ribosome display. This result indicates that VH/κ-ribosome- mRNA complex is well maintained during the selection process and shows that the antibody gene can be specifically selected by interaction between VH/κ antibodies displayed ribosome complexes and antigen.
Fig. 8. Binding activity and selectivity of 3D8 VH/ê through ribosome display. A.
ELISA of translation mixtures. Each reaction condition was showed as a table. Translated mixtures with 3D8 VH/ê RNA (1, 3), without RNA (2), and 3D8 VH/ê RNA with cycloheximide (4), were reacted with ssDNA (1, 2, and 4) or BSA (3). B. Selection and amplification of 3D8 gene by RT-PCR. Translated mixture with 3D8 RNAs (lane 1, 3), distilled water (lane 2), and 3D8 RNAs with cycloheximide (lane 4), were selected with ssDNA (lane 1, 2, and 4) or BSA (lane3).
1 2 3 4
Antigen ssDNA ssDNA - ssDNA 3D8 VH/ê
RNA + - + +
Cycloheximide - - - +
1 2 3 4
Antigen ssDNA ssDNA - ssDNA 3D8 VH/ê
RNA + - + +
Cycloheximide - - - +
B. TP-specific scFv selection by using ribosome display
1. Immunization of mice
Blood samples from TP-peptide immunized mice were taken 7 days after the fourth boost and antibody production was determined by ELISA. The ELISA results from the mouse sera that had highest titer presented as a graph (Fig. 9). Splenic cells were isolated from the spleen immunized mouse that showed the highest antibody titer.
2. PCR amplification and preparation of mRNA
VH and kappa-chain DNA were amplified by RT-PCR and assembled into VH/ê-DNA fragments using the (Gly4Ser)3 linker sequence, upstream T7/back primer and downstream Ck/for primer (Fig. 10). The T7/back primer contained a T7 promoter and ribosome binding site and the Ck/for primer had no stop codon. The Ck region of kappa chain acts as a spacer that tethers the synthesized protein to the ribosome and helps proper folding of scFv. Assembled VH/ê chain DNAs of 1.1 kb was used for preparation of mRNA.
0 0.2 0.4 0.6 0.8 1 1.2 1:5000 1:10000 1:20000 1:40000 1:80000 1:160000 s e r u m d i l u t i o n Absorbance at 405 nm
Fig. 9. Titration of immunized sera by indirect ELISA. After immunization
with TP-peptide, antibody production of immunized mouse was determined by ELISA. The ELISA results from the mouse sera that had highest titer presented. Relative antigen binding curves were plotted for immunized sera (u) and pre-immunized sera (n).
Fig. 10. Amplified DNA fragments of VH DNA, kappa chain DNA and assembled
VH/k DNA in agarose gel electrophoresis. VH DNA and kappa chain DNA were separately amplified by RT-PCR from the total RNA of lymphocytes that were obtained from a terminal protein (TP)-peptide immunized mouse. After gel purification, VH DNA and kappa chain DNA were assembled with (Gly4Ser)3 linker DNA by PCR. A. Purified VH and kappa chain DNA. Lane 1: 1kb plus DNA marker, Lane 2: 340 bp VH DNA, Lane 3: purified VH DNA, Lane 4: 750 bp scFv, Lane 5: kappa chain DNA. B. Assembled VH/ê DNA library. Lane 1: 1 kb plus DNA marker, Lane 2: 1.2 kb-sized assembled scFv DNA library.
A. 1 kb ▶ B. 1 2 1 kb ▶ 1 2 3 4 5
3. In vitro transcription and translation
The prepared VH/κ-DNA library was used to transcribe in vitro RNA with T7 RNA polymerase. The transcribed mRNA revealed a single band of an expected size by gel electrophoresis. After the synthesized mRNA was translated by a Flexi rabbit reticulocyte lysate system, the translation mixture was subjected to SDS-PAGE followed by autoradiography (Fig. 11). The translated products were detected as two major band s of about 35 kDa and 45 kDa. The translated product with luciferase mRNA had a radioactive band of 61 kDa in size. The unexpected 35kDa protein might be dead end product resulted from stop codon within library DNA.
Luciferase, 61 kDa Library proteins 45 kDa ▶
1 2 3
30 kDa ▶ 20 kDa ▶Fig. 11. In vitro translated library proteins detected by autoradiography. In vitro
translation was carried out with in vitro transcribed mRNA. After in vitro translation with [35S] labeled methionine, the products of the translation reaction were separated by SDS-PAGE and visualized by autoradiography. Lane 1: [14C] methylated protein molecular weight markers, Lane 2: synthesized library proteins, Lane 3: protein translated with luciferase mRNA as control
4. Selection of TP-specific scFv gene
Microtiter plates were used for affinity selection. After selection, eluted mRNA was amplified by RT-PCR to confirm the specificity of synthesized ARM complex against TP-peptide. A single clear DNA band was detected only from translation mixture selected against TP-peptide. No band was observed from PBS-coated wells or when untranslated mixture (translated for 0 min) was used (Fig. 12). This result indicated that TP-peptide-specific scFvs genes could be selected against immobilized TP-peptide antigen by ribosome display.
During four rounds of selection, nested PCR primers such that primers for the next round that those for the previous round, i.e., D2 in the second, D3 in the third, and D4 in the fourth round were used. Thus, the recovered DNA became progressively shorter in each cycle. The recovered band was clearer when an inner 3’ end primer was used rather than the same primer (Fig. 13). To confirm TP-specific selection, a negative control reaction was included for each selection process. No band was recovered from the PBS-coated wells in any round of selection. However, a recovered band was observed in the non-translation control of the third round of selection. This might be because the translation reaction was not completely blocked. The selected DNA pools were analyzed by radioimmunoassay to verify the enrichment of binding activity to TP-peptide (Fig. 14). Luciferase was used as a negative control. Progressive enrichment was observed by RIA and the binding activity of after 3 rounds of selection was about five times higher than the negative
Fig. 12. Selection and amplification of VH/ê gene against TP -peptide.
Confirmation of the specificity against TP-peptide was conducted under various conditions. After selection, eluted RNA was amplified by RT-PCR and the RT-PCR products were analyzed by agarose gel electrophoresis. This result ind icates that selection of scFv that binds to TP-peptide was successfully and the library DNA could be amplified by their specific interaction. Lane 1: translation mixture selected from a TP-peptide-coated well. Lane 2: translation mixture selected from a PBS-coated well. Lane 3: untranslated mRNA of VH/ê. Lane 4: RT-PCR with elution buffer.
M 1 2 3 4
Fig. 13. Effect of internal primer in RT-PCR. Two different primers were used for
RT-PCR. The amplification was much better using D4, internal primer (lane 2) than D3, 3’ primer (lane 1). The reaction with Distilled water (lane 3, lane 4) by these primers was used as negative control. This result indicates that the use of an internal primer increases a sensitivity and specificity of RT-PCR.
Fig. 14. Analysis of enriched TP -peptide -specific scFv pools. Recovered DNAs
from each round of ribosome display were used to analyze the presence of specific binders. Similar quantities of DNA from each round were used for in vitro transcription/translation. The [35S] methionine labeled in vitro translated protein was bind to TP-peptide, and analyzed by RIA. Luciferase protein was translated as a negative control (-). 1st, 2nd, and 3rd selected pool are marked as 1st, 2nd, and 3rd.
0 500 1000 1500 2000 2500 3000 (-) 1st 2nd 3rd t r a n s l a t e d p r o t e i n s cpm
5. Soluble scFv production and sequence analysis
The DNA selected after the fourth round was inserted into the expression vector, pCANTAB5E. Approximately 150 colonies were isolated and their respective soluble proteins were expressed. The periplasmic extracts from individual colonies were tested for production of antigen-specific scFvs by indirect ELISA (Fig. 15). Several clones showed binding activity to TP-peptide. Among these clones, the four clones which exhibit the highest immunoreactivity with TP-peptide were isolated. ELISA results showed that the isolated scFv clones also have binding activity to the functional human HBV DNA polymerase expressed by baculovirus expression system (Fig. 16).
The expression of each scFv was confirmed by Western blot analysis using anti-E tag antibodies (Fig. 17). Proteins of approximately 35 kDa in size were expressed from each of the four selected clones. The deduced amino acid sequences of these four scFvs are shown in Fig. 18. Using the DNA sequences, I designated the VH and Vk ge ne families of the clones based on Werner Müller’s database (http://www.dnaplot.de/input/mouse_v.html). The heavy chain of clones G6 and F7 belo nged to the VH1 gene family, that of clone G2 belonged to the VH3 gene family and that of the D10 clone belonged to the VH5 gene family. The light chain of clone G6 belonged to the Vk 21 group III subgroup, that of clone G2 belonged to the Vk 4/5 group IV subgroup and that of clones F7 and D10 belonged to the Vk 9B group V subgroup. Sequencing alignment using the Vector NTI software program showed
that the light chains of the F7 and D10 clones have very similar sequences (97%). The linker sequence had some mutations, but the se were mostly silent mutations or did not influence protein structure to any great extent presumably. This result indicates that random mutation is introduced and intact scFv structures are selected by ribosome display.
Fig. 15. Isolation of scFvs after 4th selection. The DNA selected after the fourth
round was inserted into the expression vector, pCANTAB5E. Approximately 150 colonies were isolated and the periplasmic extracts from individual colonies were tested for production of antigen-specific scFvs by indirect ELISA. Several clones have binding activity to TP-peptide.
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 1 14 27 40 53 66 79 92 105 118 131 144
Clones from 4th selection
Absorbance at 405 nm
Fig. 16. Binding activity of selected anti-TP scFvs. After the 4th selection, selected
scFv DNAs were cloned into the pCANTAB5E vector. The periplasmic extracts from each clone were tested for binding activity against TP-peptide and HBV DNA polymerase by ELISA. Full length HBV DNA polymerase (FPL-pol), TP-peptide, bovine serum albumin (BSA) and PBS were used to test the binding activity.
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1 G6 G2 F7 D10 clone Absorbance at 405 nm
G6 G2 F7 D10 M
Fig. 17. Detection of the expressed anti-TP scFvs by Western blot. Four
clones designated as F7, D10, G6 and G2, that had the highest signal in ELISA were selected and their expressions were confirmed by Western blotting. M: size marker, scFv protein size is arrowed.
47 83 32.5 6.5 16.5 25 kDa 175
Fig. 18. Alignment of the amino acid sequence of selected TP-specific scFvs. CDRs and (Gly4Ser)3 linker worked as box. The identical and conserved sequences in black and gray respectively are indicated. The labeling of CDRs was performed according to standard antibody engineering protocols.43
IV. DISCUSSION
In this study, the slightly modified ribosome display using eukaryotic translation system was established. Anti-DNA antibody, 3D8 was specifically selected against ssDNA as a model system. The selected 3D8 RNAs sequences from translation complexes were recovered by RT-PCR. By applying this model system, TP-specific scFvs were selected and enriched from the immunized mouse library. Four TP-specific scFv clones were selected and their sequences were analyzed. These four scFvs showed intact scFv structure and specific binding activity to TP-peptide; three of them, the G6, G2, and D10 clones, also bound functional human HBV DNA polymerase in an ELISA. These selected anti- TP scFvs could be used in the future to further understand HBV replication.
In vitro display techniques have some potential advantages over in vivo
display. These include ease of generation of larger libraries; fewer biases caused by cellular expression; and more facile application of round-by-round mutagenesis. Ribosome display, one of the in vitro display methods, was achieved here using an in
vitro transcription and translation process. These processes, which were based on Escherichia coli or rabbit reticulocyte system, have been described previously.17, 20
In these reports, functional scFvs were expressed and selected by ribosome display. Since TP-peptide was poorly immunogenic, I needed a large library to obtain specific scFvs using the display method. Because the library size of a ribosome
display is not limited by transformation, antigen-specific scFvs would be more easily obtained from DNA libraries using ribosome display than by in vivo display methods. A eukaryotic method was slightly modified from that described by He et al. and Makeyev et al.20, 33 In my work, the transcription and translation steps were carried out separately and the latter being performed using a rabbit reticulocyte system. By uncoupling transcription and translation, translation without a reducing agent was possible, and this could potentially result in greater functional expression of a single-chain antibody.48 Translation using a rabbit reticulocyte system has some potential advantages over an E. coli ribosome display system in that the selection conditions are less complex due to lower RNase activity.15 Moreover, eukaryotic conditions might improve the translation and/or folding efficiency of some proteins.
Internal primers were used for the RT-PCR in this system. In principle, there should be no advantage to use an internal primer in ribosome display since mRNA is released from the complex before RT-PCR. However, the results are much better with an internal primer than with a 3' end primer (Fig. 12). If some of the ribosomes remained associated with mRNA after the dissociation procedure, this might inhibit the RT-PCR reaction. However, I treated the samples with EDTA and an RNA isolation kit to dissociate the mRNA from the ribosomes. Furthermore, little mRNA should remain associated with the ribosome after processing, as an RNA isolation kit was used originally for isolation from the cell. However, using nested PCR with internal primers seems to increase the sensitivity and specificity of repeated PCR of a library.