The draft genome sequencing for Flavobacteriaceae strain EG-1, isolated from coastal seawater in Korea, was performed using Illumina HiSeq platform. As a result, the draft genome was comprised of a total length of approximately 4.45 Mbp with G + C content of 33.5%, and included a total of 3,719 protein-coding, 7 rRNA, 37 tRNA, 4 non-coding RNA, and 14 pseudo genes. The strain EG-1 had various biopolymer degradation-related genes and carotenoid biosynthesis related genes, and is assumed to contribute to the cycling of organic matters in the marine environment.
Keywords: Flavobacteriaceae, draft genome sequence, Illumina HiSeq
The family Flavobacteriaceae is the largest bacterial group (comprising 143 genera) of the phylum Bacteroidetes. It is found in a wide variety of marine, freshwater, and soil habitats (McBride, 2014). Most of members were characterized as Gram-negative, rod-shaped, chemoheterotrophic, and carotenoid- or flexirubin-pigmented bacteria possessing menaquinone 6 as the sole respiratory quinone. The family Flavobacteriaceae was subdivided based on the 16S rRNA gene sequence-based phylogenetic analysis, among which species of “marine clade”
were known to contribute greatly to the mineralization of primary produced organic matter in various marine habitats (Kirchman, 2002). Several members of the marine clade were also known to be opportunistic pathogens such as Tenacibacculum maritimum (Suzuki et al., 2001) or potentially exacerbate existing disease (Bowman and Nowak, 2004; McBride, 2014).
In this report, the draft genome sequence and annotation of a marine Flavobacteriaceae strain EG-1 is described.
An orange-pigmented marine bacterium, designated strain EG-1, was isolated from coastal seawater, Guryongpo (35° 59' 23.3'' N, 129° 33' 56.3'' E) of Korea, using a standard dilution plating method on 10 fold-diluted marine agar 2216 (MA;
Difco). The strain EG-1 was routinely cultured on MA at 25°C for 3 days. Phylogenetic analysis based on 16S rRNA gene sequence showed that strain EG-1 belonged to the family Flavobacteriaceae and was closely related to Aurantibacter crassamenti HG732
T(98.4% sequence similarity), followed by Kriegella aquimaris KMM 3665
T(94.3% sequence similarity) and Zobellia galactanivorans DsiJ
T(93.6% sequence similarity).
The genomic DNA was extracted from cells grown in marine broth 2216 (Difco) at 25°C for 2 days using MagAttract
®HMW DNA kit (Qiagen) according to the manufacturer’s instruction.
The sequencing of the draft genome was performed on the Illumina Hiseq platform with TruSeq Nano DNA (350 bp insert
Korean Journal of Microbiology (2020) Vol. 56, No. 3, pp. 324-326 pISSN 0440-2413
DOI https://doi.org/10.7845/kjm.2020.0076 eISSN 2383-9902
Copyright ⓒ 2020, The Microbiological Society of Korea
Draft genome sequence of Flavobacteriaceae strain EG-1 isolated from coastal seawater
Ji-Sung Oh and Dong-Hyun Roh*
Department of Microbiology, Chungbuk National University, Cheongju 28644, Republic of Korea
연안 해수에서 분리된 Flavobacteriaceae 균주 EG-1의 유전체 서열분석
오지성 ・ 노동현*
충북대학교 자연과학대학 미생물학과
(Received August 13, 2020; Revised August 20, 2020; Accepted August 25, 2020)
*For correspondence. E-mail: [email protected];
Tel.: +82-43-261-3368; Fax: +82-43-264-9600
Draft genome of Flavobacteriaceae strain EG-1 ∙
325
Korean Journal of Microbiology, Vol. 56, No. 3 size) library by Macrogen Inc. The de novo assembly was
performed by SPAdes (version 3.10.0). The estimated complete- ness and contamination of genome were verified with CheckM (version 1.0.18) (Parks et al., 2015). Genome annotation was performed by the NCBI Prokaryotic Genome Annotation Pipeline (PGAP) (Tatusova et al., 2016) and additional function of the predicted genes were conducted by EggNOG 5.0 (Huerta- Cepas et al., 2018), BlastKOALA with KEGG database (Kanehisa et al., 2016) and Rapid Annotations using Subsystems Technology (RAST) server with SEED (Aziz et al., 2008).
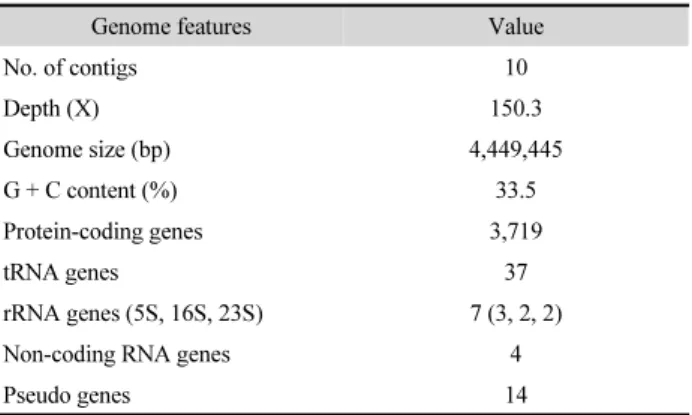
The genome statistics are shown in Table 1. The draft genome sequence of strain EG-1 consisted of a 4,449,445 bp (10 contigs; genome coverage, 150.3X) with a DNA G + C content of 33.5%. The results of CheckM estimation indicated that genome completeness at 96.6% with 0.61% contamination and 0.0% strain heterogeneity. The genome was shown to contain 3,719 protein-coding, 7 rRNA (5S, 16S, 23S), 37 tRNA, 4 non-coding RNA and 14 pseudo genes.
As a result of genome annotation and analysis of functional characterization, the draft genome sequence of strain EG-1 contained various biopolymer degradation-related genes;
xylanase Xyl, xylose isomerase XylA, xylulose kinase XylB, N-acetylglucosamine-6-phosphatase deacetylase NagA, glu- cosamine-6-phosphate deaminase NagB, type I pullulanase PulA, α-amylase AmyA and exodeoxyribonuclease III XthA. It also contained carotenoid biosynthesis related genes; 15-cis- phytoene synthase CrtB, phytoene desaturase CrtI and β- carotene 3-hydroxylase CrtZ. The genome sequence contained antibiotic-resistance related genes; tetracycline resistance protein TetA, penicillin-binding protein MrcA/MrdA, penicil- linase repressor BlaI, virginiamycin A acetyltransferase Vat,
multiple antibiotic resistance protein MarC, and multidrug efflux pump subunit AcrB. It contained bacterial secretion system-related genes such as outer membrane protein TolC, preprotein translocase subunit SecA/SecD/SecE/SecG/SecY/
YajC, YidC/Oxa1 family membrane protein insertase YidC, fused signal recognition particle receptor FtsY/SRP54, signal recognition particle subunit SRP54 and sec-independent protein translocase protein TatA/TatC. Nitrogen metabolism- related genes nitrite reductase large subunit NirB, ferredoxin- nitrate reductase NarB, nitrous oxide reductase maturation protein NosD/NosF/NosL, nitrous oxide reductase transmem- brane maturation protein NosY and nitrous-oxide reductase NosZ were also found in the genome sequence. Based on the genome analysis, strain EG-1 is thought to play an important role of the degradation of various biopolymers and the cycling of organic matters.
Nucleotide sequence accession numbers
The strain EG-1 has been deposited in Korean Culture Center of Microorganisms (KCCM) and Japan Collection of Microorganisms (JCM) under the accession number KCCM 43340 and JCM 33636, respectively. The draft genome sequence is accessible in GenBank under the accession number WJPK 00000000. The version described in this paper is version WJPK01000000.
적 요
이 연구에서는 Illumina Hiseq platform을 사용하여 연안 해 수로부터 분리된 Flavobacteriaceae 균주 EG-1의 유전체 염 기서열 해독을 수행하였다. 그 결과, 유전체는 대략 4.45 Mbp 의 길이 및 33.5%의 G + C 함량으로 구성되었고, 전체 3,719개 의 단백질 암호 유전자, 7개의 rRNA 유전자, 37개의 tRNA 유 전자, 4개의 non-coding RNA 유전자 및 14개의 위유전자 (pseudo gene)가 확인되었다. EG-1 균주는 다양한 생물 고분 자 물질 분해 관련 유전자 및 카로테노이드 생합성 관련 유전 자들을 가지고 있었고, 해양 환경에서 유기물 순환에 기여할 것으로 추정된다.
Table 1. Genome features of Flavobacteriaceae strain EG-1
Genome features Value
No. of contigs 10
Depth (X) 150.3
Genome size (bp) 4,449,445
G + C content (%) 33.5
Protein-coding genes 3,719
tRNA genes 37
rRNA genes (5S, 16S, 23S) 7 (3, 2, 2)
Non-coding RNA genes 4
Pseudo genes 14
326
∙ Oh and Roh미생물학회지 제56권 제3호
Acknowledgments
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2017R1 D1A3B04033871).
References
Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M, et al. 2008. The RAST server: rapid annotations using subsystems technology.
BMC Genomics 9, 75.
Bowman JP and Nowak B. 2004. Salmonid gill bacteria and their relationship to amoebic gill disease. J. Fish Dis. 27, 483–492.
Huerta-Cepas J, Szklarczyk D, Heller D, Hernández-Plaza A, Forslund SK, Cook H, Mende DR, Letunic I, Rattei T, Jensen LJ, et al.
2018. eggNOG 5.0: a hierarchical, functionally and phylogene- tically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 47, D309–D314.
Kanehisa M, Sato Y, and Morishima K. 2016. BlastKOALA and
GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 428, 726–731.
Kirchman DL. 2002. The ecology of Cytophaga-Flavobacteria in aquatic environments. FEMS Microbiol. Ecol. 39, 91–100.
McBride MJ. 2014. The Family Flavobacteriaceae, pp. 643–676. In Rosenberg E, DeLong EF, Lory S, Stackebrandt E, and Thompson F. (eds.), The Prokaryotes: Other Major Lineages of Bacteria and The Archaea, Springer Berlin Heidelberg, Berlin, Heidelberg, Germany.
Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, and Tyson GW.
2015. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055.
Suzuki M, Nakagawa Y, Harayama S, and Yamamoto S. 2001.
Phylogenetic analysis and taxonomic study of marine Cytophaga- like bacteria: proposal for Tenacibaculum gen. nov. with Tenaci- baculum maritimum comb. nov. and Tenacibaculum ovolyticum comb. nov., and description of Tenacibaculum mesophilum sp.
nov. and Tenacibaculum amylolyticum sp. nov. Int. J. Syst. Evol.
Microbiol. 51, 1639–1652.
Tatusova T, DiCuccio M, Badretdin A, Chetvernin V, Nawrocki EP, Zaslavsky L, Lomsadze A, Pruitt KD, Borodovsky M, and Ostell J. 2016. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 44, 6614–6624.