Draft genome sequencing of the Cytophagaceae strain SJW1- 29, isolated from the Seomjin River in Korea, was performed using the PacBio platform. The draft genome was composed of 7,065,248 bp with a G + C content of 50.7%. Genome anno- tation revealed that the genome comprised 5,684 protein-coding genes, 9 rRNA genes, 41 tRNA genes, 2 ncRNA genes, and 273 pseudo genes. Genome analysis revealed that the strain SJW1-29 harbors several carbohydrate active enzymes, which are provides insights for understanding the polysaccharide degradation system of the strain at the genome level.
Keywords: Cytophagaceae, draft genome sequence, PacBio
The family Cytophagaceae is the largest group in the phylum Bacteroidetes (the Cytophaga–Flavobacterium–Bacteroides group) (Stanier, 1940). At present, the family Cytophagaceae comprises 13 genera with validly published names (http://www.
bacterio.net/cytophagaceae.html), and Cytophaga is the type genus of the family. Members of family Cytophagaceae have been isolated from a wide variety of habitats, including fresh- water sources, ginseng, rhizopshere, glaciers, marine environments, and plants. Cytophagaceae comprises rod-shaped, non-spore
forming, frequently pigmented, chemoorganotrophic bacteria (Kirchman, 2002).
Strain SJW1-29 was isolated from a brackish water from Seomjin River, South Korea (34°58'15.8" N, 127°45'34.0" E), using a standard dilution plating method on marine agar 2216 (MA; BD Difco) after 2 weeks of culture at 20°C. The optimum temperature for growth was determined and cultures were maintained on MA at 28°C. Strain SJW1-29 was grown in media containing 1.0% (w/v) xylan from beechwood (Sigma- Aldrich) or pectin isolated from apple (Sigma-Aldrich) as the main carbohydrate. A representative sequence of the 16S ribo- somal RNA (rRNA) gene isolated from strain SJW1-29 was compared with those of other members of the family Cyto- phagaceae using EzBioCloud server (https://www.ezbiocloud.
net/). Comparison of the 16S rRNA gene sequences showed that strain SJW1-29 was most closely related to Persicitalea jodogahamensis Shu-9-SY12-35C
T(91.3%), followed by Rhab- dobacter roseus R49
T(90.6%) and Arundinibacter roseus DMA-K-7a
T(90.0%), whereas similarity to other strains of Cytophagaceae was lower than 90.0%. These values were significantly lower than the cut-off value of 95% for the allocation of a strain to a novel genus (Kim et al., 2014).
Furthermore, phylogenetic analyses of the 16S rRNA gene
Korean Journal of Microbiology (2020) Vol. 56, No. 4, pp. 422-425 pISSN 0440-2413
DOI https://doi.org/10.7845/kjm.2020.0112 eISSN 2383-9902
Copyright ⓒ 2020, The Microbiological Society of Korea
Draft genome sequence of the polysaccharide degrading bacterium Cytophagaceae strain SJW1-29
Kiwoon Baek and Ahyoung Choi*
Nakdonggang National Institute of Biological Resources (NNIBR), Sangju 37242, Republic of Korea
다당류 분해 세균인 Cytophagaceae 과의 신균 SJW1-29의 유전체 염기서열 초안
백기운 ・ 최아영*
국립낙동강생물자원관
(Received November 2, 2020; Revised December 9, 2020; Accepted December 14, 2020)
*For correspondence. E-mail: [email protected];
Tel.: +82-54-530-0722; Fax: +82-54-530-0729
Draft genome of Cytophagaceae strain SJW1-29 ∙
423
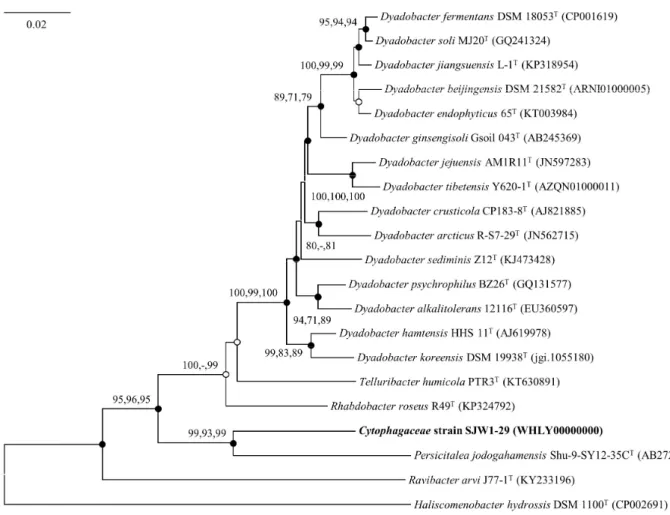
Korean Journal of Microbiology, Vol. 56, No. 4 sequence showed that strain SJW1-29 formed a distinct lineage
among members of the family Cytophagaceae (Fig. 1).
To isolate genomic DNA from strain SJW1-29, the cells were cultured in marine broth 2216 (BD Difco) at 28°C for 4 days, following which a DNeasy Blood and Tissue Kit (Qiagen) was used to extract genomic DNA. The draft genome was sequenced using the PacBio RS II platform with the SMRTbell library from DNA Link, Inc.. Genome assembly of the filtered PacBio reads (987,242,904 bp; 116,476 subreads; N
50, 11,196 bp) was performed using the Hierarchical Genome Assembly Process 3 within PacBio single-molecule, real-time analysis v2.3.0 (Chin et al., 2013). Potential contamination in genome assembles were checked by the Contamination Estimator by 16S (ContEst16S) and CheckM (Version 1.0.18) tools (Parks et
al., 2015; Lee et al., 2017). Genome annotation was performed using the NCBI Prokaryotic Genome Annotation Pipeline (Tatusova et al., 2016), and additional functions for the predicted genes were performed using the Clusters of Orthologous Group (COG) categories (Tatusov et al., 2003), BlastKOALA with the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (Kanehisa et al., 2016) and Rapid Annotations using Subsystems Technology v2.0 server (Aziz et al., 2008).
The genome statistics are summarized in Table 1. The draft genome sequence of strain SJW1-29 contained 7,065,248 bp (4 contigs; contig N
506,512,965 bp) with a G + C content of 50.7%. The results of ContEst16S and CheckM estimation indicated that this genome was not contaminated. The genome was shown to contain 5,684 protein-coding genes, 9 rRNA
Fig. 1. Neighbour-joining tree based on nearly complete 16S rRNA gene sequences showing the phylogenetic position of strain SJW1-29T with closely related taxa. Bootstrap values > 70 are indicated and based on 1,000 replications (methods: neighbour-joining, maximum likelihood, and maximum parsimony; -, no value). Filled circles indicate that the corresponding nodes were recovered by all treeing methods. Open circle indicates that the corresponding node was recovered by the neighbour-joining and maximum-parsimony methods. Haliscomenobacter hydrossis DSM 1100T (CP002691) was used as an out-group. The scale bar depicts 0.02 substitutions per nucleotide position.
424
∙ Baek and Choi미생물학회지 제56권 제4호
genes (5S, 16S, and 23S), 41 tRNA genes, 2 ncRNA genes, and 273 pseudo genes. A total of 5,317 genes were functionally assigned to categories based on COG assignments. In the genome of strain SJW1-29, the number of genes associated with general function prediction (R; 13.4%), carbohydrate transport and metabolism (G; 8.0%), and cell wall/membrane/envelope biogenesis (M; 7.6%) was more than that associated with other functions. Based on KEGG, strain SJW1-29 possessed all genes for most of the carbohydrate metabolism pathways of amino sugar and nucleotide sugar metabolism.
The genome annotations and functional characterization analysis showed that the draft genome sequence of strain SJW1-29 contained multiple genes encoding putative polysaccharide- degrading enzymes, which included eleven glucosidases (EC 3.2.1.3, EC 3.2.1.20, and EC 3.2.1.21), two chitinases (EC 3.2.1.14), four cellulases (EC 3.2.1.4), eight xylanases (EC 3.2.1.8), six amylases (EC 3.2.1.1 and 3.2.1.68), one alginate lyase (EC 4.2.2.3), and six pectinesterases (EC 3.1.1.11). These results suggest that the Cytophagaceae strain SJW1-29 can use polysaccharides for nutrition demonstrates flexible adaptation to available energy and/or carbon sources.
Nucleotide sequence accession number
The strain SJW1-29 was deposited in the Korean Collection for Type Cultures (KCTC 72493) and NITE Biological Resource Center (NBRC 114061). The draft genome sequence is accessible in GenBank under the accession number WHLY00000000.
This version of the project (01) has the accession number WHLY01000000 and comprises the sequences WHLY01000001–
WHLY01000004.
적 요
이 연구에서는 PacBio RS II platform을 사용하여 섬진강에 서 분리한 Cytophagaceae 과에 속하는 균주 SJW1-29의 유전 체 염기서열 해독을 수행하였다. 그 결과, 유전체의 총 길이는 7,065,248 bp, G + C 함량은 50.7%로 구성되었다. 전체 5,684 개의 단백질 코딩 유전자, 9개의 rRNA 유전자, 41개의 tRNA 유전자, 2개의 ncRNA 유전자 및 273개의 pseudo 유전자가 확 인되었다. 유전체 연구에 따르면 균주 SJW1-29는 여러 탄수 화물 활성효소를 보유하고 있으며, 이는 유전체 수준에서 균 주의 다당류분해 시스템을 이해하기 위한 통찰력을 제공한다.
Acknowledgments
This work was supported by a grant from the Nakdonggang National Institute of Biological Resources (NNIBR), funded by the Ministry of Environment (MOE) of the Republic of Korea (NNIBR202002102).
References
Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M, et al. 2008. The RAST server: rapid annotations using subsystems technology.
BMC Genomics 9, 75.
Chin CS, Alexander DH, Marks P, Klammer AA, Drake J, Heiner C, Clum A, Copeland A, Huddleston J, Eichler EE, et al. 2013.
Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 10, 563–569.
Kanehisa M, Sato Y, and Morishima K. 2016. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 428, 726–731.
Kim M, Oh HS, Park SC, and Chun J. 2014. Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes.
Int. J. Syst. Evol. Microbiol. 64, 346–351.
Kirchman DL. 2002. The ecology of Cytophaga-Flavobacteria in aquatic environments. FEMS Microbiol. Ecol. 39, 91–100.
Lee I, Chalita M, Ha SM, Na SI, Yoon SH, and Chun J. 2017.
ContEst16S: an algorithm that identifies contaminated prokaryotic genomes using 16S RNA gene sequences. Int. J. Syst. Evol.
Microbiol. 67, 2053–2057.
Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, and Tyson GW.
2015. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Table 1. General genomic features of strain SJW1-29
Genome features Value
No. of contigs 4
Genome size (bp) 7,065,248
N50 (bp) 6,512,965
G + C content (mol%) 50.7
Protein-coding genes 5,684
rRNA genes (5S, 16S, 23S) 9 (3, 3, 3)
tRNA genes 41
ncRNA genes 2
Pseudo genes 273
Draft genome of Cytophagaceae strain SJW1-29 ∙
425
Korean Journal of Microbiology, Vol. 56, No. 4
Res. 25, 1043–1055.Stanier RY. 1940. Studies on the Cytophagas. J. Bacteriol. 40, 619–
635.
Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV, Krylov DM, Mazumdr R, Mekhedov SL, Nikolskaya AN, et al. 2003. The COG database: an updated version includes
eukaryotes. BMC Bioinformatics 4, 41.
Tatusova T, DiCuccio M, Badretdin A, Chetvernin V, Nawrocki EP, Zaslavsky L, Lomsadze A, Pruitt KD, Borodovsky M, and Ostell J. 2016. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 44, 6614–6624.