An algicidal bacterium, designated strain Nhm2S1, was isolated from tidal flat of South Sea, Korea. Herein, we report draft genome sequence of strain Nhm2S1, which was determined using Illumina HiSeq X-ten platform. The assembled genome of strain Nhm2S1 consists of 10 contigs with a total length of 3,926,919 bp and the genomic DNA G + C content was 43.4 mol%. The draft genome encoded 3,876 protein-coding genes, 11 rRNA genes, 65 tRNA genes, 4 non-coding RNA genes and 26 pseudo genes. The genome contained genes (redP/R) involved in the biosynthesis of the algicidal pigment prodigiosin, which was thought to helpful in understanding of algae-killing properties.
Keywords: Halobacillus sp. Nhm2S1, draft genome sequence, Illumina HiSeq X-ten
The genus Halobacillus, belong to the family Bacillaceae, was first described by Spring et al. (1996) and currently comprises 21 species with validly published names (Parte et al., 2020; https://lpsn.dsmz.de/). In general, members of the genus Halobacillus were isolated from various saline environ- ments, including salt lake, saline soil and saltern, and were characterized as Gram-stain-positive, spore-forming, rod-shaped, heterotrophic and moderately halophilic bacterium (Spring et al., 1996). In this report, we describe the draft genome sequence
and annotation of an algicidal bacterium Halobacillus sp.
Nhm2S1.
An algicidal bacterium against marine diatom Skeletonema costatum, designated strain Nhm2S1, was isolated from tidal flat, South Sea of Korea. The isolate was aerobically grown on marine agar 2216 (Difco) at 25°C for 3 days. Comparison of the 16S rRNA gene sequences showed that strain Nhm2S1 was most closely related to Halobacillus faecis IGA7-4
Twith 99.9% sequence identity.
For the genome sequencing of strain Nhm2S1, the cells were incubated in marine broth 2216 (Difco) at 25°C for 2 days and the genomic DNA extraction was performed using MagAttract
®HMW DNA kit (Qiagen) according to the manufacturer’s instructions. Draft genome sequencing was performed using Illumina HiSeq X-ten platform with TruSeq Nano DNA (350 bp insert size) library by Macrogen Inc. Trimming of adapters and quality checking of sequencing data were performed by Trimmomatic (version 0.36) and FastQC (version 0.11.5), respectively. The de novo assembly of qualified reads was performed by SPAdes (version 3.13.0). Genome completeness and contamination were verified with CheckM (Version 1.0.18) (Parks et al., 2015). The genome annotation was conducted by NCBI Prokaryotic Genome Annotation Pipeline (PGAP) (Tatusova et al., 2016) and RAST tool kit (RASTtk) (Brettin et al., 2015). Additional function of the predicted genes were conducted by PATRIC (Wattam et al., 2017), EggNOG 5.0 (Huerta-Cepas
Korean Journal of Microbiology (2021) Vol. 57, No. 3, pp. 226-228 pISSN 0440-2413
DOI https://doi.org/10.7845/kjm.2021.1069 eISSN 2383-9902
Copyright ⓒ 2021, The Microbiological Society of Korea
Draft genome sequence of an algicidal bacterium Halobacillus sp.
Nhm2S1
Ji-Sung Oh and Dong-Hyun Roh*
Department of Biological Sciences and Biotechnology, Chungbuk National University, Cheongju 28644, Republic of Korea
살조성 세균 Halobacillus sp. Nhm2S1의 유전체 염기서열 분석
오지성 ・ 노동현*
충북대학교 대학원 생명시스템학과
(Received August 19, 2021; Revised August 30, 2021; Accepted September 2, 2021)
*For correspondence. E-mail: [email protected];
Tel.: +82-43-261-3368; Fax: +82-43-264-9600
Draft genome sequence of Halobacillus sp. Nhm2S1∙
227
Korean Journal of Microbiology, Vol. 57, No. 3 et al., 2018), BlastKOALA with KEGG database (Kanehisa et
al., 2016), and RAST server with SEED database (Aziz et al., 2008). Orthologous average nucleotide identity (OrthoANI) and in silico digital DNA-DNA hybridization (dDDH) were calculated using the Orthologous Average Nucleotide Identity Tool (OAT, https://www.ezbiocloud.net/tools/orthoani) (Lee et al., 2016) and Genome-to-Genome Distance Calculator (GGDC) web server version 2.1 (Meier-Kolthoff et al., 2013), respectively.
AntiSMASH 6.0 (https://antismash.secondarymetabolites.org/) (Blin et al., 2021) for secondary metabolite gene cluster, SignalIP 5.0 (http://www.cbs.dtu.dk/services/SignalP/) (Almagro Armenteros et al., 2019) for secretary proteins and dbCAN2 tool (Zhang et al., 2018) for carbohydrate-active enzymes were used to predict algicidal functions.
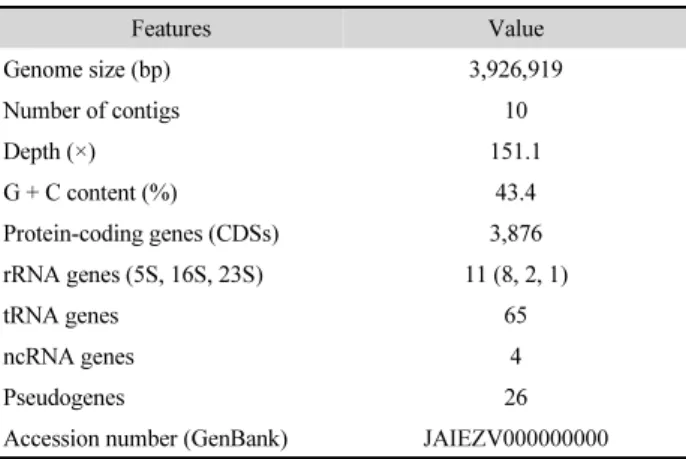
The draft genome of strain Nhm2S1 consisted of 10 contigs with a total length of 3,926,919 bp (N50 value of 1,105,605).
The sequencing depth of coverage was 151.1× and the genomic DNA G + C content was 43.4 mol%. A total of 3,876 protein coding genes, 11 rRNA genes (8 of 5S rRNA, 2 of 16S rRNA, and 1 of 23S rRNA), 65 tRNA genes, 4 non-coding RNA and 26 pseudo genes were predicted (Table 1). The results of CheckM estimation indicated that genome completeness was 99.33% with 0% contamination and 0% strain heterogeneity.
Genome of the strain Nhm2S1 was annotated using RASTtk (Brettin et al., 2015) and assigned as a strain of genus Halobacillus. OrthoANI and dDDH values between strain Nhm2S1 and H. faecis IGA7-4
Twere 94.8% and 58.8% (based on recommended formula 2, dDDH = identities /HSP length), respectively. These orthoANI and dDDH values were below the recommended threshold of 95~96% and 70%, respectively,
for species delineation (Richter and Rosselló-Móra, 2009).
The genome of strain Nhm2S1 completely encoded central carbohydrate metabolism such as glycolysis, gluconeogenesis, pyruvate oxidation, citrate cycle, pentose phosphate pathway and PRPP biosynthesis. It encoded bacterial motility related genes such as chemotaxis (cheABCDRVWXY and mcp), flagellar assembly (fliACDEFGHIJKLMNOPQRSTWY, flhABFG, flgBCDEFGKLM, motAB, flaG, and flbD) and pilus system (pilABCNMOT), which would play roles in directing and moving bacteria toward microalgae (Meyer et al., 2017). For a better understanding of the algicidal activity of strain Nhm2S1, secondary metabolites biosynthesis was investigated. Genome contained complete C5 (dxr, dxs, ispE ispG, ispH, ispDF, and idi) and C10-C20 (idi and ggpS) isoprenoid genes related terpenoid backbone biosynthesis.
Additionally, 8 secondary metabolite gene clusters, ectoine, type II PKS, beta-lactone, 2 terpenes, linear azol(in)e-containing peptide (LAP) and RiPP-like, and 2 siderophore, were found with antiSMASH 6.0. Some genes for biosynthesis of secondary metabolites (e.g. prodigiosin, piperidine, pyridine alkaloid, carotenoid, monobactam, streptomycin, novobiocin and tropane) were also found. Among them, prodigiosin was reported as an algicide pigment (Jeong et al., 2005), and the genome of strain Nhm2S1 had a redP/R gene involved in the initial step of pigment synthesis. Analysis using SignalIP 5.0 represented 361 encoding secretory proteins among the 3,876 genes. Total 84 carbohydrate-active enzymes, 30 glycoside hydrolases (GH), 22 glycosyl transferases (GT), 1 polysaccharide lyase (PL), 13 carbohydrate esterases (CE), 7 auxiliary activities (AA), and 11 carbohydrate-binding modules (CBM) were found. Of these, 15 enzymes contained signal peptide, in which two proteins (locus_tag = K2225_00205 and K2225_12825) contained signal peptide and two modules of GH and CBM.
This genome information will help understand the algae- killing properties through comparative genomics with the strain Nhm2S1 and other strains in the genus Halobacillus.
Nucleotide sequence and strain accession numbers The draft genome sequence and strain of Halobacillus sp.
Nhm2S1 has been deposited to GenBank and the Korean Culture Center of Microorganisms under the accession number JAIEZV000000000 (version JAIEZV010000000) and KCCM 43419, respectively.
Table 1. Genomic features of Halobacillus sp. Nhm2S1
Features Value
Genome size (bp) 3,926,919
Number of contigs 10
Depth (×) 151.1
G + C content (%) 43.4
Protein-coding genes (CDSs) 3,876
rRNA genes (5S, 16S, 23S) 11 (8, 2, 1)
tRNA genes 65
ncRNA genes 4
Pseudogenes 26
Accession number (GenBank) JAIEZV000000000
228
∙ Oh and Roh미생물학회지 제57권 제3호
적 요
살조성 세균 Nhm2S1 균주는 남해의 갯벌로부터 분리되었 다. 본 연구에서는 Illumuna Hiseq X-ten platform을 사용하여 Nhm2S1 균주의 유전체 서열을 수행하였다. Nhm2S1 균주의 조립된 유전체는 10개의 contig로 구성되었고, 염색체 길이는 3,926,919 bp이며 43.4 mol% G + C 함량을 지니고 있었다. 유 전체는 3,876개의 단백질 암호 유전자, 11개의 rRNA 유전자, 65개의 tRNA 유전자, 4개의 non-coding RNA 유전자 및 26 위 유전자(pseudo gene)를 암호화하였다. 게놈에는 살조성 색소 프로디지오신의 생합성에 관여하는 유전자(redP/R)가 포함 되어 있어 살조 특성을 이해하는 데 도움이 될 것이다.
Acknowledgments
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2017R1 D1A3B04033871).
Conflict of Interest
The authors have no conflict of interest to report.
References
Almagro Armenteros JJ, Tsirigos KD, Sønderby CK, Petersen TN, Winther O, Bru nak S, von Heijne G, and Nielsen H. 2019.
SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 37, 420–423.
Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M, et al. 2008. The RAST server: rapid annotations using subsystems technology.
BMC Genomics 9, 75.
Blin K, Shaw S, Kloosterman AM, Charlop-Powers Z, van Wezel GP, Medema MH, and Weber T. 2021. antiSMASH 6.0: improving cluster detection and comparison capabilities. Nucleic Acids Res. 49, W29–W35.
Brettin T, Davis JJ, Disz T, Edwards RA, Gerdes S, Olsen GJ, Olson R, Overbeek R, Parrello B, Pu sch GD, et al. 2015. RASTtk: a modular and extensible implementation of the RAST algorithmfor building custom annotation pipelines and annotating batches of
genomes. Sci. Rep. 5, 8365.
Huerta-Cepas J, Szklarczyk D, Heller D, Hernández-Plaza A, Forslund SK, Cook H, Mende DR, Letunic I, Rattei T, Jensen LJ, et al.
2018. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 47, D309–D314.
Jeong H, Yim JH, Lee C, Choi SH, Park YK, Yoon SH, Hur CG, Kang HY, Kim D, Lee HH, et al. 2005. Genomic blueprint of Hahella chejuensis, a marine microbeproducing an algicidal agent.
Nucleic Acids Res. 33, 7066–7073.
Kanehisa M, Sato Y, and Morishima K. 2016. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 428, 726–731.
Lee I, Kim YO, Park SC, and Chun J. 2016. OrthoANI: an improved algorithm and software for calculating average nucleotide identity.
Int. J. Syst. Evol. Microbiol. 66, 1100–1103.
Meier-Kolthoff JP, Auch AF, Klenk HP, and Göker M. 2013. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics 14, 60.
Meyer N, Bigalke A, Kaulfuß A, and Pohnert G. 2017. Strategies and ecological roles of algicidal bacteria. FEMS Microbiol. Rev. 41, 880–899.
Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, and Tyson GW.
2015. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055.
Parte AC, Sardà Carbasse J, Meier-Kolthoff JP, Reimer LC, and Göker M. 2020. List of Prokaryotic names with Standing in Nomenclature (LPSN) moves to the DSMZ. Int. J. Syst. Evol. Microbiol. 70, 5607–5612.
Richter M and Rosselló-Móra R. 2009. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad.
Sci. USA 106, 19126–19131.
Spring S, Ludwig W, Marquez MC, Ventosa A, and Schleifer KH.
1996. Halobacillus gen. nov., with descriptions of Halobacillus litoralis sp. nov. and Halobacillus trueperi sp. nov., and transfer of Sporosarcina halophila to Halobacillus halophilus comb. nov.
Int. J. Syst. Evol. Microbiol. 46, 492–496.
Tatusova T, DiCuccio M, Badretdin A, Chetvernin V, Nawrocki EP, Zaslavsky L, Lomsadze A, Pruitt KD, Borodovsky M, and Ostell J. 2016. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 44, 6614–6624.
Wattam AR, Davis JJ, Assaf R, Boisvert S, Brettin T, Bun C, Conrad N, Dietrich EM, Disz T, Gabbard JL, et al. 2017. Improvements to PATRIC, the all-bacterial bioinformatics database and analysis resource center. Nucleic Acids Res. 45, D535–D542.
Zhang H, Yohe T, Huang L, Entwistle S, Wu P, Yang Z, Busk PK, Xu Y, and Yin Y. 2018. dbCAN2: a meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 46, W95–W101.