□ 원 저 □ Vol. 14, No. 2, November, 2006
1)
책임저자 : 김성환, 아주대학교 의과대학 소아과학교실 Tel : 031)219-5162, Fax : 031)219-5169 E-mail : pedkim@ajou.ac.kr
장기 추적 관찰된 사립체 유전자 A3243G 변이 환자의 임상표현형과 자연경과
아주대학교 의과대학 소아과학교실
안미선·정우철·황진순·김성환
= Abstr act =
Long-term Clinical Follow-up of Patients with A3243G Mitochondrial tRNA Mutation :
Clinical Phenotype and Natural Course
Mi Sun Ahn, M.D., Woo Chul Jeoung, M.D.
Jin Soon Hwang, M.D. and Sung Hwan Kim, M.D.
Department of Pediatrics, School of Medicine, Ajou University Suwon, Korea
Purpose : Mitochondrial disorder is a progressive disease, but there are no specific treatment modalities to prevent the progression. Also, there has been little understanding on the pattern of disease progression nor natural history. The aim of this study was to elucidate the initial clinical phenotypes, patterns of the disease progression, and its natural history of the patients with mitochondrial A3243G mutation.
Methods : Among the patients with biochemically or genetically confirmed mitochondrial disorders, 7 patients with A3243G mutation were included in a 7 year follow-up obser- vation(range: 3-11 years). We classified the patients into two groups by the initial clinical presentations : systemic and neurologic onset. They were clinically evaluated with serial brain MRI and MRS for the evaluation of the disease evolution patterns.
Results : The clinical manifestations of mitochondrial A3243G mutation were extremely variable; seizure, headache, dementia, myopathy, sensorineural hearing loss, external ophtha- lmoplegia, diabetes mellitus, cardiomyopathy, easy fatigability, and short stature. Among the 7 patients, 4 patients initially presented neurologic symptom such as seizure(3) and headache(1), and 3 patients systemic symptoms such as DM(2) and easy fatigability(1).
All the patients with neurologic onset showed relentless progression with recurrent stroke- like episodes and intractable seizures, and finally fell into be functionally dependent states or death. All the patients with systemic onset showed clinically silent periods for 3-10 years, and still they were in functionally independent states despite subsequent neurologic symptoms.
Conclusion : We could find out the relationship between initial clinical phenotypes and final outcomes in mitochondrial A3243G mutation. However, the population is small in this study so that a larger scaled analysis is needed.
Key Words : Mitochondrial disorder, A3243G mutation, Initial symptom, Natural history
서 론
1962년 사립체 질환이 처음 기술된 이래
1)임상 적으로 다양한 사립체 질환이 보고되고 있다. 산화 인산화(oxidative phosphorylation) 과정을 통해서 에너지 생성에 관여하는 사립체 호흡사슬은 5개의 호흡연쇄효소 복합체로 구성되어 있기 때문에 사립 체 호흡연쇄효소 결핍 질환은 생화학적으로 다양하 며, 다른 효소와 달리 호흡연쇄효소 복합체는 핵 DNA에 의해서 생성되는 단백질에 사립체 DNA에 의해서 생성되는 단백질이 결합되어서 만들어지기 때문에 유전학적 다양성을 보인다
2). 전체 환자의 75-90%는 핵 DNA 유전자 변이, 나머지 10-25%
는 사립체 DNA 유전자 변이에 의해서 다양한 형 태의 사립체 질환이 발생하는 것으로 보고되고 있 다
3-5).
사립체 유전자 tRNA Leu(UUR)을 코드화 하는 MTTL1 유전자의 3,243번째 nucleotide가 adenine 에서 guanine으로 치환되는 점돌연변이(A3243G로 표기)가 가장 흔하게 발견되는 사립체 유전자 변이 로 사립체 질환 환자의 22.3%, MELAS(mitochon- drial encephalomyopathy, lactic acidosis, and stroke-like episodes)환자의 45.1%에서 A3243G 유전자 변이가 발견된다
6). 사립체 유전자 A3243G 변이는 MELAS, Kearns-Sayre 증후군, MERRF (myoclonus epilepsy and ragged red fibers), 당 뇨, 감각신경성 난청, 말초 신경병증, 편두통, 진행 성 바깥눈근육마비(progressive external ophtha- lomoplegia), 간질 및 자폐증 같은 신경계 증상부 터 저신장증, 신 증후군, 주기성 구토와 MINGIE(mi- tochondrial neurogastointestinal encephalomyo- pathy) 같은 전신 증상까지 대단히 다양한 임상양 상으로 나타난다
3, 7). 임상증상은 소아연령부터 주 로 빠르게 진행하는 신경계 증상으로 나타나기 시 작하고, 성인에서는 주로 내분비계를 포함한 전신 증상으로 나타난다
8-10).
사립체 질환의 근본적인 치료법은 없지만 coen- zyme Q
10, dichloroacetate, thiamine, vitamin C
와 L-arginine 같이 사립체에서 에너지 대사에 보 조적 역할을 하는 약물들이 사립체 질환의 치료에 사용되고 있고, 이러한 약물들은 뇌졸증양 발작이 나 젖산혈증 같은 특정 증상을 완화시킬 수 있지 만 질병의 진행은 막지 못하고 있는 실정이다
11-13). MELAS 등 사립체 질환에서 질병이 나타나서 사 망에 이르기까지 질병의 자연경과에 대한 체계적인 연구가 거의 없기 때문에 증상이 시작된 후 어떤 방향으로 진행되는지 예상하기 어렵다. 따라서 자 연경과에 대한 이해는 사립체 질환의 조기 진단 및 치료 전략 수립에 도움을 줄 수 있다.
이에 저자들은 현재까지 근본적인 치료법이 없 는 사립체 유전자 tRNA의 A3243G 유전자 변이 가 있는 환자들에서 초기 임상유형을 조사하고, 초 기 증상에 따른 질병의 진행 양상과 예후를 장기 간 추적 관찰함으로써 A3243G 유전자 변이로 인 한 사립체 질환의 자연경과를 알아보기 위해서 본 연구를 시행하였다.
대상 및 방법
1999년부터 2005년까지 아주대학교병원 소아신 경과에 방문한 환자들 중 뇌 자기공명영상, 근전도, 신경전도속도, 뇌파 같은 신경계 검사에서 뇌병증 의 원인이 규명되지 않고 혈액 및 소변 아미노산 분석 검사, 소변 유기산 분석 검사, 지방산 분석 검사에서 대사질환의 증거가 없는 환자에서 혈액과 뇌척수액 젖산 농도 검사, 광학현미경이나 전자현 미경을 이용한 형태학적 검사와 사립체 호흡연쇄효 소 활성도 검사를 시행하여서 사립체 질환의 여부 를 진단하였다. 사립체 호흡연쇄효소 활성도 검사 는 피부 생검을 통해서 얻어진 피부 조직을 배양 하여서 확립된 피부 섬유아세포에서 시행하였다.
사립체 호흡연쇄효소 활성도 검사로 사립체 질환으
로 진단된 환자와 사립체 뇌병증, 근병증, 젖산혈
증, 반복되는 뇌졸증양 발작, 경련발작 및 편두통
으로 구성된 MELAS의 임상 진단 기준
1)을 만족
하는 환자들의 혈액에서 사립체 유전자를 추출하여
서 제한효소로 처리한 후 중합효소연쇄반응법을 이
용하여 사립체 유전자 tRNA의 A3243G 유전자 변이가 발견된 환자 7명을 대상으로 하였다. 이 중 3명의 환자는 신경과에서 의뢰된 성인 환자였다.
환자들의 의무기록을 통해 사립체 유전자 tRNA 의 A3243G 유전자 변이의 초기 증상과 임상 표현 형을 후향적으로 분석하였다. 환자들은 발현되는 첫 증상에 따라 신경계 증상으로 시작된 환자군과 전신 증상으로 시작된 환자군으로 분류하였고 또한 발병 시 연령에 따라서 소아청소년 환자와 성인 환자로 분류하였다. 사립체 유전자 tRNA의 A3243G 유전자 변이를 확인한 이후부터는 질병의 경과를 알기 위해서 소아신경과 또는 신경과 외래 진료소 에서 2005년 7월까지 정기적으로 추적 관찰하였다.
평균 추적관찰 기간은 7.4±3.0(범위: 3-11)년이었 다. 각 환자의 임상양상을 추적 관찰하면서 다른 장기를 침범하는 새로운 증상의 출현 여부와 신경 계 증상의 진행 양상을 알기 위해서 뇌파검사, 뇌 자기공명영상 검사와 뇌 자기공명분광법을 이용하 여 질병의 진행과정을 분석하였다. 환자의 최종 상 태는 마지막 방문할 때 환자의 자발적 운동 상태 와 고위 대뇌기능 수행 정도에 따라서 분석하였다.
결 과
1. 대상 환자의 일반적 특성
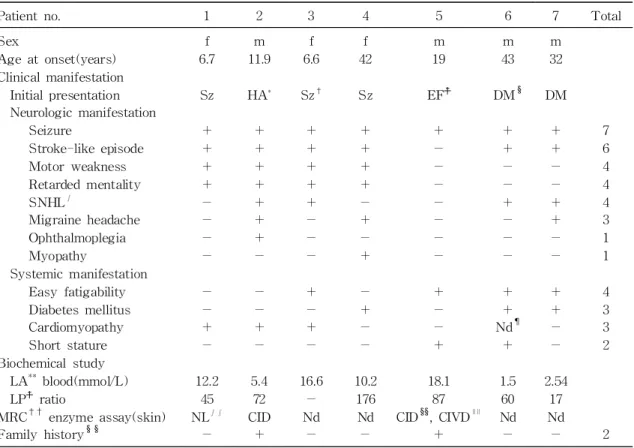
사립체 유전자 tRNA A3243G 유전자 변이가 확인된 7명의 환자 중 남자 4명, 여자 3명이었고 임상증상이 시작되는 나이는 6살에서 43살까지 다 양하였으며 4명은 사춘기 이후에 첫 증상이 시작 되었다. A3243G 유전자 변이의 첫 증상은 간질발 작(3명), 당뇨(2명), 편두통(1명), 전신 쇠약감(1명) 으로 4명의 환자는 신경계 증상, 3명의 환자는 전 신증상으로 발현되었다. 첫 증상이 전신증상으로 나타난 환자는 모두 성인 환자였고 소아 환자는 모두 신경계 증상으로 발현되었다. 추적 관찰 중 후속적으로 나타나는 A3243G 유전자 변이의 임상 양상은 매우 다양하였다. 전체 환자 7명 중 간질발 작이 가장 흔한 증상으로 모든 환자에서 발생하였 고, 뇌졸증양 발작(6명), 치매(4명), 감각신경성 청
각장애(4명), 강직성 사지마비(4명), 두통(3명), 눈 근육마비(1명), 전신 쇠약감(4명), 대사 위기(meta- bolic crisis; 3명), 심근병증(3명), 당뇨(3명), 저신 장(2명) 등이 있었다. 증례 5를 제외한 6명 환자에 서 MELAS 증후군의 특징적 소견인 뇌졸증양 발 작이 발생하였다. 이 중 증례 1과 2는 뇌졸증양 발 작이 첫 증상으로 나타났으며 나머지 4명은 추적 관찰 중 뇌졸증양 발작이 발생하였다. 첫 증상이 신경계 증상으로 나타난 환자들은 모두 반복적으로 뇌졸증양 발작이 발생하였으나, 첫 증상이 전신 증 상으로 나타난 경우는 뇌졸증양 발작이 반복적으로 나타나지는 않았다(Table 1).
생화학적 검사에서 증례 6을 제외한 모든 환자 에서 혈액 젖산 농도가 증가되었다. 이중 2명에서 만 혈액 젖산 농도가 지속적으로 증가하였고 나머 지 4명은 간헐적으로 증가하였다. 사립체 호흡연쇄 효소 활성도 검사가 시행된 3명의 환자에서 2명이 이상소견을 보였고 그 중 1명은 효소복합체 1번 결핍, 나머지 한명은 효소복합체 1번과 4번 복합결 핍을 보였다. 근전도 및 신경전달속도에 대한 검사 는 4명에서 시행하였는데 1명만 근병증이 확인되 었고 나머지 3명은 정상소견을 보였다. 가족력 조 사가 가능하였던 4명 환자의 가계에서 MELAS가 의심되는 친척이 3명 있었다. 그중 사립체 유전자 tRNA의 A3243G 유전자 변이가 확인된 사람은 증례 2번 환자의 어머니, 증례 5번 환자의 여동생 2명이었다. 친척 중 사립체 유전자 A3243G변이가 확인되었으나 MELAS의 임상양상을 나타내지 않 은 사람도 1명 있었다(Table 1).
2. 첫 증상에 따른 사립체 질환의 진행양상
환자에서 나타난 사립체 질환의 첫 증상에 따라
신경계 증상을 보인 환자군과 전신 증상을 보인
환자군으로 분류하였다. 전체 7명의 환자 중 4명
(57%)은 첫 증상이 신경계 증상으로 나타났으며 3
명(43%)은 전신 증상으로 나타났다. 첫 증상이 신
경계 증상으로 나타난 4명의 환자 중 2명은 뇌졸
증양 발작, 2명은 간질발작으로 나타났다. 그러나
첫 증상이 간질발작으로 나타난 2명의 환자 모두
뇌졸증양 발작이 발생하였다. 또한 모든 환자에서 운동실조증, 거짓연수마비와 심근병증 같은 사립체 질환의 다른 증상이 동반되었다. 추적 관찰 중 모 든 환자에서 뇌졸증양 발작이 반복적으로 발생하면 서, 후속적으로 시행한 뇌 영상검사에서 점진적으 로 미만성 대뇌피질 위축과 뇌실 확장 소견이 점 진적으로 진행하는 경과를 보였다. 또한 모든 환자 에서 간질발작은 항경련제 병합치료에도 불구하고 난치성 간질의 경과를 보였다. 최종 평가 방문시 2 명의 환자는 심한 치매와 경직성 사지마비로 인한 심한 기능장애를 보였고 증례 1은 흡인성 폐렴, 증 례 3은 비가역적인 젖산혈증으로 사망하였다(Table 2, 3).
전체 7명의 환자 중 3명(43%)은 첫 증상이 전
신 쇠약감과 당뇨 같은 전신증상으로 나타났다. 이 집단의 모든 환자는 첫 증상이 나타나고 3년에서 10년까지 다른 증상이 속발되지 않는 임상적 휴지 기를 보였다. 그러나 장기간 추적관찰 시 모든 환 자에서 뇌졸증양 발작, 간질발작, 감각신경성 난청 같은 신경계 증상이 동반되었다. 그러나 첫 증상이 신경계 증상으로 발현된 환자들과 달리 뇌졸증양 발작은 반복적으로 발생하지 않았고 후속적으로 시 행한 신경계 영상검사에서도 대뇌피질 위축과 뇌실 확장 같은 소견은 관찰되지 않았다. 최종 평가시 모든 환자들은 치매와 신체장애 없이 기능적으로 정상인 생활을 영위하고 있었다(Table 2, 3).
Table 1. Clinical Features of Patients with A3243G Mitochondrial tRNA Mutation
Patient no. 1 2 3 4 5 6 7 Total
Sex
Age at onset(years) Clinical manifestation
Initial presentation Neurologic manifestation
Seizure
Stroke-like episode Motor weakness Retarded mentality SNHL∫
Migraine headache Ophthalmoplegia Myopathy
Systemic manifestation Easy fatigability Diabetes mellitus Cardiomyopathy Short stature Biochemical study
LA**blood(mmol/L) LP☨ ratio
MRC†† enzyme assay(skin) Family history§§
f 6.7
Sz
+
+
+
+
-
-
-
-
-
-
+
-
12.2 45 NL∫∫
-
m 11.9
HA*
+
+
+
+
+
+
+
-
-
-
+
-
5.4 72 CID
+ f 6.6
Sz†
+
+
+
+
+
-
-
-
+
-
+
-
16.6
- Nd
- f 42
Sz
+
+
+
+
-
+
-
+
-
+
-
-
10.2 176 Nd
-
m 19
EF☨
+
-
-
-
-
-
-
-
+
-
-
+
18.1 87 CID§§, CIVD∥∥
+
m 43
DM§
+
+
-
-
+
-
-
-
+
+ Nd¶
+
1.5 60 Nd
- m 32
DM
+
+
-
-
+
+
-
-
+
+
-
-
2.54 17 Nd
- 7 6 4 4 4 3 1 1
4 3 3 2
2
*HA : headache, †Sz : seizure, ☨EF : easy fatigability,∫SNHL : sensory neural hearing loss,§DM : diabetes melli- tus, ∥Nd : not done, ¶LA : lactic acid, **LP ratio : lactoc acid/pyruvic acid ratio, ††NL : normal,☨☨CID : Complex I deficiency, ∫∫CIVD : Complex IV deficiency
§§mitochondrial A3243G mutation
3. 발병연령에 따른 사립체 질환의 진행양상 전체 7명의 환자 중 3명(43%)은 소아연령에서, 4명(57%)은 사춘기 이후에 첫 증상이 발현되었다.
소아연령에서 첫 증상이 발현된 환자는 모두 신경 계 증상으로 시작되었으나 사춘기 이후 첫 증상이 나타나는 경우는 4명 중 3명에서 전신증상으로 발 현되었다. 소아연령에서 증상이 나타난 환자는 모 두 반복적인 뇌졸증양 발작과 난치성 간질의 경과 를 보이면서 끊임없이 진행하여서 치매와 심한 사
지마비로 이행되었다. 그러나 사춘기 이후에 첫 증 상이 나타난 환자들 중 신경계 증상으로 발현된 증례 4를 제외하고는 모두 전신증상으로 발현되고 장기간 다른 증상 없이 휴지기를 보이다 뇌졸증양 발작, 간질발작 및 감각신경성 난청 같은 신경계 침범을 보이나 기능 장애 없이 정상적인 생활을 하고 있다(Table 2).
Table 3. Sequential Changes of Brain MRI/CT Findings in the Patients with A3243G Mitochondrial tRNA Mutation
Case Onset of symptom During follow up At final visit
SLLs* DA† VM☨ Recurrent SLLs SLLs DA VM Mental Status 1
2 3 4 5 6 7
+
+
-
-
-
-
-
-
-
-
-
-
+
+
+
+ Nd Nd
-
+
+
+
+
-
+
+
+
+
+
+
-
-
-
+
+
+
+
-
-
-
dementia dementia dementia§ dementia
normal normal normal Nd∫
Nd Nd
*SLLs : stroke-like lesions, †DA : diffuse cortical atrophy, ☨VM, ventriculomegaly, ∫Nd : not done
§patient 3 expired 6 month later after final visit
Table 2. Evolution of Clinical Manifestations in Patients with A3243G Mitochondrial tRNA Mutation
Case No. 1 2 3 4 5 6 7
Age of onset(years) Initial presentation Initial symptom
Silent phase Years(if yes) Progression
New symptoms Intractable seizures Recurrent SLLs☨☨
Final outcome
Duration of F/U(years) Dementia
Physical dependency
6.7 CNS*
Sz☨
-
CM¶
+
+
6
+ E§§
12 CNS HA∫
-
ataxia
+
+
6.8
+ D∫∫
6.6 CNS
Sz
-
CM
+
+
5.6
+ E
42 CNS
Sz
-
PBP**
+
+
10
+ D
19 S† EF§
+ 3 Sz
-
-
3
- I
43 S DM∥
+ 8 HL††, Sz, SLL
-
-
8
- I∥∥
32 S DM
+ 10 Sz, HL, SLL
-
-
11
- I
*CNS : central nervous system, †S : systemic onset, ☨Sz : seizure, ∫HA : headache, §EF : easy fatigability,
∥DM : diabetes mellitus, ¶CM : cardiomyopathy, **PBP : pseudobulbar palsy, ††HL, sensorineural hearing loss, ☨☨SLLs, stroke-like episodes, ∫∫D : functionally dependent, §§E : expired, ∥∥I, functionally inde- pendent
고 찰
사립체는 세포 생존에 필요한 에너지를 생성하 는 역할을 하는 세포 소기관으로 인체 내 모든 장 기와 조직에 존재한다. 사립체 질환은 에너지 의존 도가 높은 중추신경계, 근육 및 심장에 주로 질병 이 발생하지만 인체 내 모든 장기와 조직을 침범할 수 있기 때문에 임상증상이 대단히 다양하다
1, 2, 14). 사립체 기능 장애를 초래하는 질환은 carnitine결 핍과 같은 기질 운반 장애, 피루빈산이나 지방산 대사 이상과 같은 기질 이용 장애, Kreb 회로 장 애, 호흡 사슬 기능 장애, 산화-인산화 공역 장애 로 나누어 생각할 수 있다
14). 그러나 일반적으로 유전성 사립체 호흡연쇄효소 결핍으로 인해 사립체 기능 장애가 초래되는 경우를 사립체 질환으로 정 의하고 있다. 사립체는 자체적으로 유전자를 갖고 있는 유일한 세포 소기관이기 때문에 핵 유전자 뿐 만 아니라 사립체 유전자의 변이로 인해서도 사립 체 에너지 생성에 결함을 초래하는 사립체 질환이 발생한다. 또한 사립체 유전자는 모계유전, 이형질 성, 복제분리, 역표현, 높은 변이율, 나이에 따른 변 이경향 등 매우 독특한 유전적 특성을 가지고 있어 서 대단히 다양한 임상 표현형으로 나타난다
15-17). 사립체 질환에서 발견되는 사립체 유전자 변이로 유전자 결실, 삽입, 점 돌연변이 등 여러 가지 형태 의 유전자 돌연변이가 발견되는데
14, 18)이 중 사립 체 유전자 A3243G변이가 가장 흔히 발견되는 유 전자 변이로 성인 10만 명에 16명 정도 발견되고 있다
19). 최근 사립체 질환이 확인된 환자를 대상으 로 조사한 국내 연구에서도 사립체 유전자 A3243G 변이가 전체 환자의 22.3%에서 발견되었 으며, MELAS 환자의 45.1%에서 A3243G변이가 발견되었다
20). 국외 대규모 연구에서도 MELAS 환자의 75%에서 사립체 A3243G유전자 변이가 확 인되어 사립체 질환의 원인으로 A3243G변이가 많 은 빈도를 차지함을 알 수 있다
21-23).
사립체 A3243G 유전자 변이는 MELAS, PEO, Leigh 증후군, MERRF, Kearns Sayre 증후군, 치
매, 감각신경성 난청, 말초 신경병증, 편두통, 간질, 자폐증, 저신장증, MIDD(maternally inherited di- abetes and deafness), 신증후군, 주기성 구토와 MINGIE(mitochondrial neurogastointestinal en- cephalomyopathy), 비대심장근육병증 등 대단히 다양한 임상표현형으로 발현된다
3, 7, 17, 24-28). 본 연
구에서도 위와 같이 매우 다양한 임상증상을 보였
다. 사립체 A3243G 유전자 변이의 임상표현형 중
MELAS가 가장 흔하지만, 발병 연령에 따라 나타
나는 첫 증상이 달라 소아는 주로 뇌졸증양 발작,
간질발작 및 두통 같은 신경계 증상으로 시작되나,
성인에서는 당뇨, 저신장증, 전신쇠약감 같은 전신
증상으로 시작되는 경우가 많다
9, 10, 17). 또한 소아
환자의 30% 이상에서 심근병증, 전신쇠약감, 발육
부전 같은 전신 증상도 사립체 질환의 중요한 증
상으로 나타난다
14, 18). 위와 같은 다양한 증상들이
질환마다 단독으로 나타날 수도 있지만 시간이 지
나면서 서로 중복되어 나타나기 때문에, 본 연구에
서는 사립체 A3243G 유전자 변이가 있는 환자를
첫 증상에 따라서 신경계 발병형과 전신 발병형으
로 분류한 후 장기간 추적 관찰하였다. 이러한 시
간의 경과에 따른 증상 발현의 다양성과 관련된
예가 보고되어 있다. 8세 남자 환자가 첫 증상이
안검하수와 백내장으로 나타난 후에 뇌졸증양 발작
이 발생하여서 Kearns-Sayre 증후군으로 진단되
었지만, 환자가 14살에 사망하면서 얻은 간과 심장
근육 조직에서 사립체 유전자 A3243G변이가 발견
되어서 MELAS로 진단되었다
29). 처음에는 두통만
호소하다 점차 반맹, 정신이상, 실어증 등의 증상
을 보이다 뇌졸증양 발작이 나타나서 MELAS로
진단된 경우도 있었다
30). 또한 MIDD와 MELAS
는 사립체 A3243G유전자 변이로 생기는 다른 임
상표현형이지만, 당뇨이외에 MIDD에서 흔히 볼
수 있는 저신장증, 저체중, 감각신경성 청력소실
같은 증상들은 MELAS에서도 발견되는 증상들이
다. 따라서 당뇨, 저신장증, 심근병증 등 전신 증상
으로 발현된 환자라도 시간이 지난 후 사립체
A3243G 유전자 변이의 특징적인 신경계 증상이
후속적으로 나타날 수 있기 때문에 장기간 자연경
과를 추적 관찰하여야 진단의 오류를 피할 수 있 고 예후를 정확히 예측할 수 있다.
현재 사립체 질환의 진행을 막을 수 있는 근본 적인 치료법은 없고 단지 coenzyme Q
10과 dichlo- racetic acid 같은 호흡사슬의 생화학적 기능에 보 조적 역할을 하는 대증적 치료로 젖산 혈증 같은 증상을 호전시킬 수 있다는 몇 몇 연구자들의 단편 적이고 제한적인 보고들
11-13)이 있을 뿐이다. 그러 나 많은 수의 환자를 대상으로 장기간 임상증상의 호전이나 질병의 진행을 막는 약물의 효과를 과학 적인 방법으로 입증한 전향적 연구결과들은 아직 거의 없는 실정이다. 따라서 저자들의 연구와 같이 coenzyme Q
10을 투여하고 관찰한 질병의 경과를 자연경과로 보아도 무방하다고 판단할 수 있다
31). 또한 사립체 유전자 A3243G 변이의 자연경과에 대한 단편적인 단기 관찰 보고는 조금 있으나, 많 은 수의 환자를 대상으로 장기간 관찰한 보고는 거 의 없는 실정이다
13). 저자들은 사립체 A3243G 유전 자 변이를 첫 증상에 따라서 신경계 발병형과 전신 발병형으로 분류하고 장기간 임상증상의 경과와 예 후를 관찰함으로서 사립체 A3243G 유전자 변이의 자연 경과를 분석하였다. 신경계 증상으로 발현된 환자들은 모두 임상증상의 휴지기가 없이 간질발작 과 뇌졸증양 발작이 지속적으로 반복되면서 끊임없 이 병변이 진행하여 사망하거나 기능적으로 의존적 인 상태로 이행되었다. 반면 전신 증상으로 발현된 환자들은 모두 오랜 기간 다른 증상이 발현되지 않 는 임상적 휴지기가 있다가 나중에 간질발작 또는 뇌졸증양 발작 같은 신경계 증상이 속발되었다. 그 러나 간질발작은 항경련제로 잘 조절되었고 뇌졸증 양 발작은 반복되지 않았으며 모든 환자는 일상생 활을 영위하는데 기능적으로 장애가 없었다. 위의 결과는 첫 증상이 신경계 증상으로 발현된 환자들 에서 반복적으로 시행한 뇌 자기공명영촬영과 뇌 자기공명분광법 검사에서 미만성 뇌위축과 뇌실 확 장증이 점진적으로 진행되는 소견으로 보아서 뇌졸 증양 발작과 난치성 간질발작이 반복되면서 발생하 는 신경세포 손상으로 해석할 수 있다. 따라서 반 복되는 뇌졸증양 발작과 난치성 간질발작에 의해서
초래되는 결과는 사립체 질환의 세포 손상 기전으 로 이미 널리 알려진 에너지 불균형, 자유 라디칼 에 의한 손상과 세포사멸에 의한 것으로 생각된다
14)
. 그러나 이러한 변화가 첫 증상이 전신 증상으 로 발현된 사립체 A3243G 유전자 변이가 있는 환 자에서도 적용될지는 임상적 휴지기 후에 뇌졸증양 발작이나 간질발작 같은 신경계 증상이 발생하고 나서 임상경과를 관찰한 기간이 짧기 때문에 오랜 동안 추적관찰이 필요할 것이다.
본 연구에서 저자들은 사립체 A3243G 유전자 변이 환자에서 발병 시 첫 증상에 따라 질병의 자 연경과에 따른 예후를 예측 가능할 것으로 생각한 다. 신경계 증상으로 첫 증상이 발현된 경우는 뇌 졸증양 발작과 간질발작이 끊임없이 반복되면서 독 립적 생활이 불가능하거나 사망하는 불량한 예후를 시사하고, 첫 증상이 전신증상인 경우 장기간 임상 적 휴지기가 있으며 나중에 모든 환자에서 신경계 증상이 나타나고 예후가 양호할 것을 시사한다. 저 자들의 결과는 소수의 환자를 대상으로 이루어진 연구이기 때문에 향후 많은 수의 환자를 대상으로 한 연구가 필요할 것이다.
요 약
목 적 : 사립체 호흡연쇄효소 결핍으로 인해 발 생하는 사립체 질환은 현재 병인이나 자연경과가 자세히 밝혀져 있지 않은 진행성 질병이다. 그러나 질병의 진행을 막을 수 있는 근본적인 치료법은 없고 단지 효과가 입증되지 않은 대증치료에만 의 존하고 있는 실정이다. 저자들은 사립체 A3243G유 전자 변이가 확인된 환자에서 A3243G 유전자 변 이의 첫 임상표현형, 첫 증상에 따른 질병의 진행 양상, 예후를 파악하여 A3243G 유전자 변이의 자 연경과를 평가하고자 하였다.
방 법 : 1998-2005년까지 아주대병원 소아신경과
를 방문한 환자 중 사립체 A3243G 유전자 변이가
확인된 환자 7명을 대상으로 7년간 장기 추적 관
찰하였다(7.4±3.0 : 3-11년). 첫 증상에 따라서 대
상 환자를 신경계 증상 환자군과 전신 증상 환자
군으로 분류하였고, 첫 증상에 따른 질병의 진행양 상을 파악하기 위해서 연속적인 뇌 자기공명영상 검사와 뇌 자기공명분광법을 시행하였다.
결 과 : A3243G 유전자 변이의 임상증상은 간질 발작, 두통, 치매, 근병증, 강직성 사지마비, 눈근육 마비, 감각신경성 청각장애, 전신 쇠약감, 심근병증, 당뇨, 저신장 등 대단히 다양하다. 대상 환자 7명 중 4명은 간질발작과 두통 같은 신경계 증상으로 첫 증상으로 발현되었고, 3명은 당뇨와 전신 쇄약 같은 전신증상으로 발현되었다. 신경계 증상으로 발현된 환자들은 난치성 간질과 뇌졸중양 발작이 반복되면서 끊임없이 진행하여서 독립적인 생활이 불가능하거나 사망하였다. 전신 증상으로 발현된 환자들은 3-11년 동안 임상적 휴지기를 보이다 뇌 졸중양 발작과 간질발작 같은 신경계 증상이 동반 되었으나, 뇌졸중양 발작은 반복되지 않았고 모두 독립적인 생활이 가능하였다.
결 론 : 사립체 A3243G 유전자 변이는 첫 증상 에 따라 예후가 달라 신경계 증상으로 첫 증상이 나타나는 경우 예후가 불량하여서 뇌졸중양 발작이 끊임없이 반복되면서 사망하거나 독립적인 생활이 불가능하였다. 그러나 첫 증상이 전신증상으로 나 타나는 경우는 장기간 임상적 휴지기를 보이다 신 경계 증상이 나타나지만 예후가 양호하다. 향후 많 은 수의 환자를 대상으로 사립체 A3243G 유전자 변이의 진행양상과 자연경과를 전향적으로 규명할 필요가 있다.
References
1) Munnich A and Rustin P. Clinical spectrum and diagnosis of mitochondrial disorders. Am J Med Genet 2001;104:4-17.
2) Zeviani M and Di Donato. Mitochondrial dis- orders. Brain 2004;127:2153-72.
3) Naviaux RK. Developing a systematic approach to the diagnosis and classification of mitochon- drial disease. Mitochondrion 2004;4:351-61.
4) Chinnery PF, Turnbull DM. Mitochondrial me- dicine. QJM 1997;90:657-67.
5) Thornburn DR, Smeitink J. Diagnosis of mito-
chondrial disoclinical and biochemical approach.
J Inherit Metab Dis 2001;24:312-6.
6) Chae JH, Hwang H, Lim BC, Cheong HI, Hwang YS, Kim KJ. Clinical features of A3243G mitochondrial tRNA mutation. Brain Dev 2004;26:459-62.
7) Majamaa-Voltti KAM, Winqvist S, Remes AM, Tolonen U, Pyhtinen J, Uimonen S et al. A 3 year clinical follow-up of adult patients with 3243A>G in mitochondrila DNA. Neurology 2006;66:1470-5.
8) Walker UA, Collins S, Byrne E. Respiratory chain encephalomyopathies: A diagnostic clas- sification. Eur Neurol 1996;36:260-7.
9) Komlosi K, Bene J, Havasi V, Tihanyi M, Herczegfalvi A,Moser J, et al. Phenotypic va- riants of A3243G mitochondrial DNA mutation in a Hungarian family. Orv Hetil 2004;145:
1805-9.
10) Li JY, Kong KW, Chang MH, Cheung SC, Lee HC, Pang CY, Wei YH. MELAS syndrome associated with a tandem duplication in the D- loop of mitochondrial DNA. Acta Neurol Scand 1996;93:450-5.
11) Mori M, Yamagata T, Goto T, Saito S, Mo- mori MY. Dichloroacetate treatment for mito- chondrial cytopathy: long-term effects in MELAS. Brain Dev 2004;26:453-8.
12) Kubota M, Sakakihara Y, Mori M, Yamagata T, Momoi-Yoshida M. Beneficial effect of L-arginine for stroke-like episode in MELAS.
Brain Dev 2004;26:481-3.
13) Kubota M, Sakakihara Y, Mori M, Yamagata T, Momoi-Yoshida M. Beneficial effect of L- arginine for stroke-like episode in MELAS.
Brain Dev 2004;26:481-3.
14) De Vivo DC. The expanding clinical spectrum of mitochondrial diseases. Brain Dev 1993;15:
1-22.
15) McKenzie M, Liolitsa D, Hanna MG. Mitocho- ndrial disease: Mutation and mechanisms. Ne- urochem Res 2004;3:589-600.
16) Serra G, Picccinnu R, Tondi M, Muntoni F, Zeviani M, Mastropaolo C. Clinical and EEG findings in eleven patients affected by mito- chondrial encephalomyopathy with MERRF- MELAS overlap. Brain Dev 1996;18:185-91.
17) Maciej P, Jolanta S-C, Hanna M, Katarzyna T, Elzbieta K, Katarzyna I, et al. Diversity of
clinical symptoms in A 3243G mitochondrial DNA mutation(MELAS syndrome mutation).
Med Sci Monit 2002;8:CR767-73.
18) Bernier FP, Boneh A, Dennett X, Chow CW, Cleary MA, Thorburn DR. Diagnostic criteria for respiratory chain disorders in adults and children. Neurology 2002;59:1406-11.
19) Majamaa K, Moilanen JS, Uimonen S, Remes AM, Salmela PI, Karppa M, et al. Epidemiology of A3243G, the mutation for mitochondrial en- cephalomyopathy, lactic acidosis, and strokelike episodes: Prevalence of the mutation in an adult population. Am J Hum Genet 1998;63:
447-54.
20) Chae JH, Hwang H, Lim BC, Cheong HI, Hwang YS, Kim KJ. Clinical features of A3243G mitochondrial tRNA mutation. Brain Dev 2004;26:459-62.
21) Ciafaloni E, Ricci E, Shanske S, Moraes CT, Silvestri G, Hirano M, et al. MELAS: clinical features, biochemistry, and molecular genetics.
Ann Neurol 1992;31:391-8.
22) Goto Y. MELAS(mitochondrial myopathy, ence- phalopathy, lactic acidosis, and stroke-like episodes): clinical features and mitochondrial DNA mutation. Muscle Nerve 1995;Suppl 3:
S107-12.
23) Hammans SR, Sweeney MG, Hanna MG, Brockington M, Morgan-Hughes JA, Harding AE. The mitochondrial DNA transfer RNA- Leu(UUR) A→G(3243) mutation. A clinical and genetic study. Brain 1995;118(Pt 3):721-34.
24) Ueki I, Koga Y, Akita Y, Nishioka J, Yatsuga S, Fukiyama R, et al. Mitochondrial tRNA
gene mutations in patients having mitochondrial disease with lactic acidosis. Mitochondrion 2006;6:29-36.
25) Takeshima T, Nakashima K. MIDD and MELAS : A clinical spectrum. Intern Med 2005;44:276- 7.
26) Morovvati S, Nakagawa M, Sato Y, Hamada K, Higuchi I, Osame M. Phenotypes and mi- tochondrial DNA substitutions in families with A3243G mutation. Acta Neurol Scand 2002;
106:104-8.
27) Torroni A, Campos Y, Rengo C, Sellitto D, Achilli A, Magri C, et al. Mitochondrial DNA Haplogroups do not play a role in the variable phenotypic presentation of the A3243G mutation.
Am J Hum Genet 2003;72:1005-12.
28) Yasuotoshi K, Yukihiro A, Naoko T, Yoshihiro S, Hirohisa K. Heterogeneous presentation in A3243G mutation in the mitochondrial tRNAleu (UUR) gene. Arch Dis Child 2000;82:407-11.
29) Terauchi A, Tamagawa K, Morimatsu Y, Kobayashi M, Sano T, Yoda S. An autopsy case of mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes(MELAS) with a point mutation of mitochondrial DNA.
Brain Dev 1996;18:224-9.
30) Takahiro I, Fumihiko S, Shinichi K, Norihiro S. Slowly progressive spread of the stroke-like lesions in MELAS. Neurology 2003;61:1238-44.
31) Berbel-Garcia A, Barbera-Farre JR, Etessam JP, Salio AM, Cabello A, Gutierrez-Rivas E, et al. Coenzyme Q10 improves lactic acidosis, strokelike episodes, and epilepsy in a patient with MELAS(mitochondrial myopathy, encep-
halopathy, lactic acidosis, and strokelike epi- sodes). Clin Neuropharmacol 2004;27:187-91.