Vol. 69, No. 3, March 2019, pp. 227∼233 http://dx.doi.org/10.3938/NPSM.69.227
First-Principles Study on Temperature-Dependent Phase Transitions of Sn Polymorphs
Seong Min Jang · Chul Hong Park
∗Department of Physics Education, Pusan National University, Busan 46241, Korea (Received 3 January 2019 : revised 22 January 2019 : accepted 22 January 2019)
Based on first-principles electronic structure calculations, we estimated the variations in the thermodynamic free energies of lattice vibrations with temperature for various polymorph structures of Sn: we examined the α and β polymorphs, two kinds of hexagonal structures, through which we are able to investigate their thermal-induced phase- transition characteristics. Similar to the experimental results, a phase transition from alpha-Sn to beta-Sn was observed with increasing temperature, because the entropy of beta-Sn is larger than that of alpha-Sn. Thus, the entropy effect of phonons appears to have played an important role in the phase transition. The hexagonal structure can be more stable than the beta structure at a high temperature, but the transition temperature was higher than the melting temperature of Sn; thus, hexagonal Sn is thought to be a metastable phase.
PACS numbers: 63.20.D-, 63.20.dk, 63.70.+h
Keywords: Sn, Phase transition, Entropy, First-principles calculation, Phonon
Sn 다형체의 온도에 따른 구조 상전이 현상에 대한 제일원리 연구
장성민 · 박철홍
∗부산대학교 물리교육학과, 부산 46241, 대한민국
(2019년 1월 3일 받음, 2019년 1월 22일 수정본 받음, 2019년 1월 22일 게재 확정)
제일원리 전자구조 계산을 통하여, Sn의 다형체 구조 alpha 와 beta 구조, 그리고 hexagonal구조의 온도에 따른 안정성과 상전이 현상의 열역학적 특성 연구하였다. 전자에너지와 포논의 열에너지와 엔트로 피를 포함하는 자유에너지를 합성하여, 온도에 따른 총에너지 변화를 비교하여, 온도에 따른 구조 상전이를 계산하였다. 계산 결과는 alpha구조에서 beta구조로 상전이가 일어날 수 있는 것을 보여 주었다. 하지만, 최근 박막성장 기술로 제작된 hexagonal(H) 구조의 자유에너지를 이미 알려진 alpha구조 및 beta구조의 총에너지와 비교한 결과, Sn의 융해 온도 보다 높은 온도에서 beta구조에서 H구조로 상전이가 일어날 수 있음을 보였다. 즉, 본 계산 결과는 H가 준안정상태임을 보여 준다.
PACS numbers: 63.20.D-, 63.20.dk, 63.70.+h Keywords: Sn, 상전이, 엔트로피, 제일원리계산, 포논
∗E-mail: [email protected]
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
I. 서 론
Sn의 밧데리 음극전극 응용에 대한 연구의 관심이 매우 높다 [1]. 기존 탄소보다 축전용량이 높게 나오기 때문이다.
또한 최근 Sn으로 구성되며, 그래핀과 같은 원자구조를 가 지는 2차원 물질 Stanene이 위상부도체의 성질을 가질 수 있다고 알려져 관심이 높다 [2]. Sn은 다형체로써 잘 알려져 있다. 여러가지 원자구조를 가질 수 있어 [3,4], 온도에 따른 구조 상전이, 압력에 따른 구조 상전이가 일어나는 것이 잘 알려 져 있다. 따라서, 그 상전이 특성에 대한 실험적 및 이론적 연구가 많이 이루어져 왔다 [3–9]. Sn은 다이 아몬드형 정방구조를 가지는 비금속성의 α-Sn(회색 주석) 와 tetragonal 구조를 가지는 금속성의 β-Sn(백색 주석) 의 구조가 대표적이다. 후자의 경우 전극으로 사용된다. 한편, 286 K 이하에서는 α-Sn가 β-Sn보다 안정하고, 그 위로는 β-Sn가 안정하다고 알려져 있다. 따라서 온도를 내리면, β → α의 구조상전이가 발생한다 [2–4]. 구조적으로 α-Sn 는 β-Sn보다 쉽게 가루가 되기 쉽다. 우리 주변에서 흔히 보는 Sn은 내구성이 비교적 강한 금속의 β-Sn이다.
Sn에 대한 상전이는 압력에 의한 것과 온도변화에 의한 것 두가지 종류가 있다. Sn은 압력 변화에 따라 구조 상전이가 일어나는 것이 실험적으로 잘 알려져 있다. 압력에 대한 Sn 의 다형체의 상전이에 대한 이론적 연구는 비교적 오래전에 이루어 졌다 [4–6], Cheong과 Chang은 압력에 따라 Sn의 구조 상전이에 대해 제일 원리 계산을 통해 연구하였다, 압력 을 높여감에 따라 Sn의 구조는 α→ β → bct → bcc → hcp 구조의 상전이가 나타남을 부피변화에 따른 제일원리 총에 너지 계산을 통해, 실제 실험 결과와 유산한 상전이 특성을 성공적으로 보였다 [4].
Sn은 상온 근처에서 온도 변화에 따른 상전이는 전극으 로써 응용에 문제를 발생시킨다. 금속성 β-Sn 구조가 반 도체상 α-구조로 바뀔 때, 체적이 약 30% 정도 증가한다.
이것은 Sn-전극을 사용하는 소자의 성능 저하를 일으킨다.
따라서, 온도에 따른 상전이 연구가 중요하게 다루어 졌다.
온도에 따른 다형체의 상전이에 대해 이론적 연구는, 격자 진동에너지를 온도에 따라 계산하여, α 에서 β 구조 사이 구조 상전이가 총에너지 관점에서 이해할 수 있다는 것을 Na와 Park이 보였다 [7]. 최근 Hörmann et al.은 DFT계 산을 수행하여, 표면효과 때문에, 나노크기에서 Sn에 대해 상전이온도가 크기 낮아질 수 있음을 DFT계산을 이용하여 보였다 [8].
최근 Sn에 대한 흥미로운 연구 결과로써, 박막 성장 기 술을 적용하여, 기존에 없었던, hexagonal 구조의 Sn을 성 공적으로 만들었다. 그래핀과 유사한 원자구조를 가지는 hexagonal 구조의 Sn의 경우, 위상 물질로써 가능성 때문에
Fig. 1. (Color online) The atomic structure of polytypes:
(a) α-Sn, (b) β-Sn, (c) Hexagonal-Sn (H-Sn), and (d) Graphite-Sn (G-Sn).
큰 관심을 불러일으키고 있다 [2]. Shao와 Beaufils는 박막 성장 기술을 통해, Sn을 육각격자 (Hexagonal, H) 구조로 만드는데 성공하였다 [9]. 위상 전자구조 특성을 비롯한 특이한 성질을 가지는 것으로 알려져 있어, 연구의 관심이 매우 높다 [2]. Saxena et al.은 HRT EM 을 사용한 구조 적 특성 분석을 통해, 박막기술을 이용하여 성장한 Sn에서, U V−V is 광스펙트럼 측정과 제일원리 계산의 비교를 통해, 육각대칭 격자의 성장을 확인하였다 [10]. 현재, hexagonal 구조가 박막기술로 성장 되었으나, 구조의 열적 안정성에 대한 이해는 잘 알려져 있지 않다. 이 구조가 열역학적 안 정성에 대한 연구가 필요하다.
따라서, 본 연구에서는 제일원리 전자구조 계산법을 이 용하여, Sn의 다형체구조의 상대적 안정성을 연구하였다.
특히 기존에 알려진 α와 β 구조의 Sn과 함께 hexagonal(육 각)구조의 Sn이 안정되게 존재할 수 있는 가능성을 탐색하 였다. 제일 원리 총에너지 계산법을 이용하여, Sn의 α 와 β 상 및 hexagonal (이하 H) 구조, graphite (이하 G) 구조에 대해 절대 0도에서 안정성을 비교하였고, 또한 온도에 따른 격자진동의 열에너지와 헬름홀츠자유에너지를 계산하여, 온도에 대한 α, β, H, G 구조의 상대적 안정성을 조사하 였다. 연구결과 구조의 Sn은 고온에서 안정될 수 있으나, 실제로 구현하는 것은 매우 어려운 것을 알게 되었다.
Table 1. The calculated electronic energies of Sn polytypes at ground state are shown, relatively to that of the α-Sn.
The unit is eV/atom. The calculated lattice constants are compared with the experimental data.
Sn-poly types Electronic energy Lattice constants (Å)
Lattice structrure
(Eel) Presnt (a, c) Exp. (a, c)
α 0 6.66 6.491 [20] cubic
β 0.014 5.87, 3.10 5.820, 3.175 [20] T etragonal
H 0.018 3.00, 2.83 - , 3.3 [5] Hexagonal
G 0.284 5.13, 6.00 - Graphite
Table 2. The position of k-points, which are indicated at the band structures in Fig. 2 and Fig. 4, are described at reciprocal lattice structure within the Brillouine zone.
Sn-poly types ×b1 ×b2 ×b3 ×b1 ×b2 ×b3
α 0 0 0 Γ 0.5 0.25 0.75 W
0.5 0 0.5 X 0.5 0.5 0.5 L
β 0 0 0 Γ -0.5 0.5 0.5 M
0 0 0.5 X 0.32 0.32 -0.32 Z
H, G 0 0 0 Γ 0.33 0.33 0 K
0.5 0 0 M 0 0 0.5 A
II. 연구 방법
밀도함수이론 (DFT) 에 [11,12] 기반한 제일원리 전자구 조와 총에너지 계산을 project augmented wave 슈도포텐 셜방법을 이용하여 [13], V ASP (5.4.4) 프로그램을 이용하 여 수행하였다 [14]. 그리고 격자진동의 포논 계산을 VASP 와 접목한 Phonopy(1.11.2.80) 프로그램을 이용하여 수행 하였다 [15]. 전자에너지 계산 시 브릴로앙영역 내 적분을 위한 kpoints는 Monkhorst-Pack [16] 방법을 이용하여 만 들었고 Γ− point 를 중심에 두었다. 그리드 N × N × N 의 N값을 높여가며 수렴하는 에너지값을 구조마다 구하였다.
파동함수의 평면파 전개를 위한 에너지 상한값을 Sn은 241 eV를 권장하고 있으나 본 연구에서는 이보다 더 큰 450 eV 를 이용하여 시험하였다. 격자진동의 양자 포논의 에너지 띠구조를 기본셀을 확장시켜 만든 Supercell(슈퍼셀)을 이 용하여 계산하였다. 그리고 포논 계산은 dynamical matrix 를 이용하여 계산되는데, 이때, 한 원자의 변위가 이웃 원 자에 미치는 힘을 고려하기 때문에, 슈퍼셀을 사용해야 한 다. 그리고 슈퍼셀 내 원자 수가 많을수록 정확한 계산이 된다. 기본적으로 개 정도의 원자 수가 있어야 신뢰 가능한 포논에너지를 계산할 수 있으므로 최소 개로 설정하였다.
제일원리총에너지 계산을 통해 전자에너지를 계산하였고, Phonopy를 이용하여 온도변화에 따른 포논의 헬름홀츠자 유에너지(G)를 합하여 계의 총에너지를 계산하였다. 한편 Sn은 semi-core상태로써 4d를 가전도대에 포함하여 계산하 였는데, 이때 Sn-4d궤도가 국소궤도를 형성하므로, 이 궤도
에 대하여 local density approximation (LDA)근사가 적절 치 아니하여 generalized gradient approximation (GGA) 으로 밀도변화율을 이용하여, 보정하였으며, 강하게 국소 화된 Sn-4d궤도에서 self-interaction효과 오차를 보정하기 위해 LDA+U방법을 이용하였다 [17,18]. 이때 적절한 U 값을 찾는 것이 중요한데, U값을 변화시키면서, 잘 알려져 있는 α → β 의 상전이 온도가 실험에서 관측된 286 K가 되도록 맞추었다. 이때 U값은 3.5 eV이었다. (아래에 자세 한 논의 있음) U값을 감소시키면, 상전이 온도가 높아지게 계산되는데, 예로써, U값을 2.0 eV로 하면, 상전이 온도가 417 K가 되었다. 이 U = 3.5eV을 H 와 G구조에 대해서도 동일하여 적용하여 안정성을 비교하였다.
포논에 의한 열 에너지는 U =
∫
dωDph(ω) ¯hωph
exp(¯hωph/τ )− 1 (1) 로 계산할 수 있다. 여기서 Dph(ω)의 포논상태밀도이다.
Bose-Einstein 통계를 적용하였다. 부피가 일정하다고 가 정하고, 포논에 의한 Helmholtz Free Energy(F)는 다음과 같이 주어진다.
F (T ) =
∫ ∞
0
dωDph(ω) ln [
2 sinh (β¯hω
2 )]
= U− T S (2) 식 (2)를 계산하여, 온도에 따른 다형체의 상대적 안정성을 계산하였다. S 는 열역학적 엔트로피 (entropy, S) 와 U 는 내부에너지이다. 엔트로피는 부피가 일정할 때,
S =−(∂F
∂T )
V
(3)
Fig. 2. The electronic strucrure of Sn polytypes: (a) α-Sn, (b) β-Sn, (c) H-Sn, and (d) G-Sn.
로 구할 수 있다. S 를 다음 식을 이용하여 계산할 수 있다.
S(T ) = −1 N
∑
k,i
kBln[
1− exp(
−hνk,i
kBT )]
+1 N
∑
k,i
kB
hνk,i
kBT [
exp(hνk,i
kBT
)− 1]−1 (4)
이다. 위 식은 포논에 대한 Bose-Einstein 통계를 적용한 것이고, 전자인 경우 Fermi-Dirac 통계를 적용하여 유사한 식으로 엔트로피를 구할 수 있다. 엔트로피는 격자진동에 너지가 낮은 물질에서 그 효과가 크다.
III. 계산 결과 및 논의
본 연구에서는 총 네 가지 구조인 α, β, H, G 구조에 대해 온도에 따른 열적 안정성을 비교하였다. 이들의 격자 구조는 각각, fcc, bct, hexagonal, hexagonal 구조이다. 각 상의 원자구조는 Fig. 4에 보여준다. 각각 기본셀 내 2, 4, 1, 4개의 원자를 가진다. G 구조와 H 구조는 hexagonal 기 둥형 격자구조를 가지는데 원자가 적층되는 순서는 H 구조 는 원자위에 원자가 위치하며 적층하는 반면 Fig. 4(a), G-
구조에서는 ABABA 순서로 적층한다 (Fig. 4(d)). 여기서 실선은 기본셀 격자벡타를 나타낸다.
(전자에너지 계산)
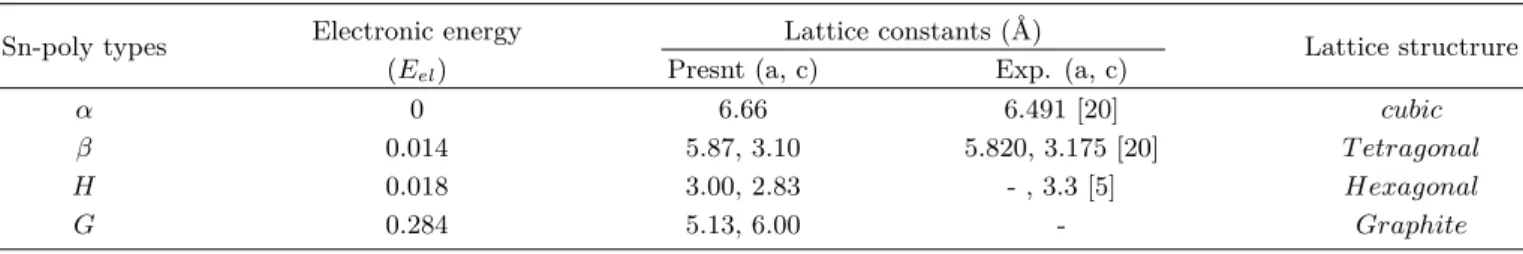
Sn 내 전자의 파동함수를 평면파 전개하였는데, 이를 위 한 최대평면파에너지를 300 eV로부터 증가시키면서 비교한 결과 450 eV정도에서 수렴하였다. 따라서 본 계산은 최대 평면파 에너지로 450 eV을 사용하였다. 역격자 k-공간의 블 릴로앙 영역에서 그리드 점들을 이용하여 k-점 세트를 만들 어 계산을 진행하였다. k-그리드 수를 변화시키면서, 전자 총에너지의 변화를 조사하여 수렴되는 값을 찾았다. 에너지 값의 상대오차 값이 0.002% 이내로 즉 1 meV/atom 내로 충분히 수렴될 때 electron energy를 계산에 적용하였다. 계 산된 전자총에너지 비교 결과와 격자상수 계산 값이 Table 1에서 주어 진다. α 구조는 F CC 셀에서 Γ−X−W −Γ−L, β 구조는 BCT 격자구조에서 : Γ− X − M − Γ − Z, H 및 G 구조는 H 구조의 격자에서 Γ− M − K − Γ − A의 경로로 하여 계산하였다. 각 구조의 K 점의 좌표는 Table 2와 같다. 각 구조의 원자 당 전자 에너지는 Table 1에서 보인 바와 같이, α < β < H < G 구조 순서대로 에너지가 커졌다.
Fig. 3. (Color online) The density of electronic states in Sn-polytypes: (a) α-Sn, (b) β-Sn, (c) H-Sn, and (d) G-Sn.
Fig. 4. (Color online) The band structure of phonon energy states of Sn-polytypes: (a) α-Sn, (b) β-Sn, (c) H-Sn, and (d) G-Sn. The rights show the denisties of phonon states.
Fig. 2는 Sn 다형체의 전자의 에너지 띠 구조를 보여 준다.
Fig. 3는 전자상태 밀도와 원자 각운동량 궤도에 투사된 밀 도를 보여준다. α-Sn은 semi-metal(반금속)임을 보여준다.
그리고 다른 구조의 Sn은 금속의 성질을 보여 준다. 그리고 β-Sn과 H-Sn에서는 금속성질을 보여준다. 페르미준위에 서 전자상태 밀도가 높아, 전기 전도성이 높은 금속 성질을 가지는 것으로 보인다. 약하게나마 sp 혼성 결합이 α, β, H 구조에서는 존재하는 것으로 보인다. α-Sn에서 페르미준위 아래 약 6 eV 근처에서 강한 sp 혼성 결합을 보인다. 이 sp- 혼성에너지가 다른 구조의 Sn에 비해 낮다. 이것은 강한 sp 혼성상호 작용을 말해 주는데, 이것에 의해 valence band 와 coduction band가 분리되는 것을 볼 수 있다. 이러한 이유로 가장 낮은 전자 에너지를 가지는 것으로 해석된다.
다른 구조에서 sp 혼성 결합 상호작용은 유사한 듯하다.
G-Sn에서 특이한 것은 s 오비탈이 모두 채워진 후 p 오 비탈이 채워지는 것으로 나타난다. 이 때문에 G 구조의 에 너지가 다른 구조보다 높게 형성되는 것으로 보인다. 높은 전자 밀도가 형성되는 에너지 값을 비교하면 β 구조가 H 구조보다 더 아래쪽에 형성되고 있음을 알 수 있으며, 이 때 문에 β 구조가 H 구조보다 더 낮은 전자에너지를 형성하는 것으로 해석된다.
(격자진동 에너지 계산)
격자진동이 양자화된 포논의 헬름홀츠 자유에너지를 계 산하기 위해 기본셀을 확장하여 슈퍼셀을 만들었다. α-Sn 와 β-Sn는 3× 3 × 3, H-Sn는 4 × 4 × 4, G-Sn은 4 × 4 × 1로 확장하여 V ASP 을 통해 Force Constant matrix를 계산한 후 Phonopy code 를 이용하여 포논의 열역학적 에너지를 구하였다. Fig. 4는 계산된 포논의 에너지 띠 구조를 보여 준다. α 구조는 5.4 THz, β 구조는 3.8 THz, H 구조는 4.2 THz, G 구조는 4.6 THz의 포논 모드의 최대 진동수를 가진 다. G 구조에서 최대 -1.7 THz 정도까지 음의 진동에너지의 포논 상태가 있는 것처럼 계산된다. 이것은 G-Sn구조에서 원자변위 의한 에너지 변화가 비조화적(an-harmonic)임을 말해 준다, G-구조의 열역학 에너지와 엔트로피를 계산할 때, 음의 에너지에 대한 상태밀도를 양의 에너지에 대응하 는 것으로 전환 합성하여 계산하였다. 이 방법은 정확한 방법은 아니나 상태밀도를 버릴 수 없기 때문에 에너지의 절대값을 취하여 상태밀도를 합성하여 계산하였다.
음의 에너지를 가진 포논상태가 계산되는 것은 G 구조 가 불안정한 구조임을 말해 준다. 즉 유사한 전자구조를 가지는 C이나 Si 물질과 달리 Sn의 경우에는 G 구조가 준 안정상태로도 존재할 수 없다는 것을 말해 준다. C과 달리 Sn은 d-궤도가 활성적이고 원자 결합에 참여한다. 따라서 ABAB적층 구조의 G 구조에서 원자 층간에 Sn-Sn상호 작 용이 있어, 판상 구조의 G 구조가 불안정해 지는 반면, AA 적층구조의 단순한 H 구조는 준안정 상태로 존재한다.
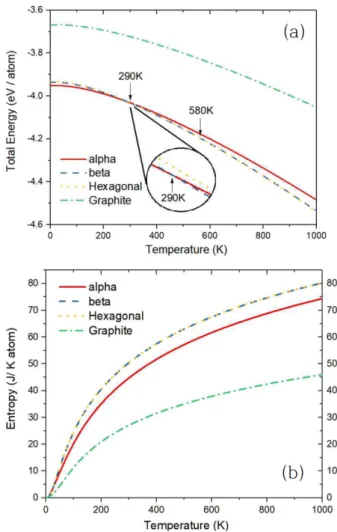
Fig. 5. (Color online) (a) The summation of the Helmholtz free energy of phonon and the electronic en- ergy are shown, depending on temperature and (b) the entropy depending on temperaure are shown.
(온도에 따른 구조 상전이)
이제 온도가 변화에 따라 Sn의 각 구조의 열적 안정성을 논의한다. 계의 에너지는 크게 격자진동에 의한 격자진동의 헬름홀츠자유에너지와 원자와 전자 간 상호작용에너지 및 전자-전자 상호작용에너지를 고려한 전자에너지를 합하여, 총에너지를 계산하였다. 포논의 자유에너지의 온도에 따른 변화를 고려하여, 온도에 따른 상전이 및 각 구조의 열적 안정성을 시험하였다.
Fig. 5(a) 은 Sn 다형체의 전자에너지와 포논의 자유에 너지 F 를 합한 값을 온도에 따라 그린 것이다. T = 0K 일 때 원자당 0점-양자에너지는 α = -3.951 eV, β = - 3.936 eV, H = -3.932 eV, G = -3.667 eV로 계산되었다.
α < β < H < G의 순이다. F − T 그래프에서 기울기는 엔트로피를 나타낸다. Fig. 5(b) 는 온도에 따른 엔트로피 값을 보여준다. 대략 엔트로피의 크기는 H > β > α > G 의 순서이다. 이 결과는 온도가 증가함에 따라 식 (4) 에
의해 더 큰 entropy를 가지는 β 또는 H 가 α 구조보다 더 안정해 질 가능성이 있다는 것을 의미한다.
T = 0 K일 때, α < β < H < G 의 순서대로 에너지가 분포한다. 이후 293 K가 되면, β < α < H < G 의 순서로 에너지가 바뀌고, 560 K일 때, H < β < α < G 의 순서로 에너지가 바뀐다. 290 K, 580 K에서 각각 α → β → H 의 상전이가 일어남을 알 수 있다. 즉 고온에서 H 구조가 안정해 질수 있음을 보여 준다. 하지만 H 구조가 생성되는 온도는 Sn의 융해 온도 505 K보다 높은 온도로써, 단순하 게 온도를 올리는 것으로는 고체상태의 Sn에서 H 구조는 나타나지 않는다. 박막 성장 기술로 인위적으로 만들어진 H상은 [9,10] 준안정 상태임을 말해 준다.
더 정확한 계산을 위해서는 전자의 열적 자유에너지와 엔트로피를 모두 고려해 주어야 한다. 하지만, 0 K에서 는 β 와 H 구조의 전자의 자유에너지의 상대적 값은 2.47 meV/atom이고, 500 K에서는 2.62 meV/atom로 변화는 0.15 meV/atom에 불과하다. 즉 전자의 열적 자유에너지 및 엔트로피가 상전이에 미치는 영향은 매우 미비하여, 고 려할 필요가 없는 것을 알았다.
IV. 결 론
본 연구에서는 Sn 다형체들의 열역학적 안정성에 대해 연구하였다. α, β 구조 뿐만 아니라 hexagonal (육각대칭, H) 구조나 그레파이트 (graphite, G) 구조를 포함하여, 총 네 가지의 Sn 다형체 구조에 대해 제일원리 계산을 통해, 각 원자구조의 열역학적 안정성에 대해 연구하였다. 절대 영도에서 제로점 양자효과에너지는 α < β < H < G 의 순서이었다. 온도에 따른 전자에너지 + 격자진동에너지를 합한 총에너지의 변화를 추적한 결과, 290 K, 580 K에서 각각 α → β → H 의 상전이가 일어남을 알 수 있었다.
즉 고온에서 H 구조가 안정해질 수 있음을 보여 준다.
하지만 H 구조가 생성되는 온도는 Sn의 융해 온도 505 K 보다 높은 온도로써, 단순하게 온도를 올리는 것으로는 고체상태의 Sn에서 H 구조는 나타나지 않는다. 박막 성장 기술로 인위적으로 만들어진 H 구조상은 준안정 상태임을 말해준다.
감사의 글
이 논문은 부산대학교 기본연구지원사업 (2년) 에 의해 연구되었습니다.
REFERENCES
[1] J. W. Wang, X. H. Liu, S. X. Mao and J. Y. Huang, Nano Lett. 12, 5897 (2012).
[2] J. C. Garcia, D. B. de Lima, L. V. C. Assali, J. F.
Justo, J. Phys. Chem. C. 115, 13242 (2011).
[3] W. Paul, J. Appl. Phys. 32, 2028 (1961).
[4] G. A. Busch and R. Kebn, Solid State Commun. 11, 1 (1960).
[5] B. H. Cheong and K. J. Chang, Phys. Rev. B 44, 4103 (1991).
[6] G. Kresse and J. Hafner, Phys. Rev. B 47, 558 (1993).
[7] Y. Yao and D. D. Klug, Solid State Commun. 151, 1873 (2011).
[8] S. H. Na and C. H. Park, J. Korean Phys. Soc. 56, 498 (2010).
[9] N. G. Hörmann, A. Gross, J. Rohrer and P. Kag- hazchi, Appl. Phys. Lett. 107, 123101 (2015).
[10] J. Shao, C. Beaufils and A. N. Kolmogorov, Sci.
Rep. 6, 28369 (2016).
[11] S. Saxena, R. P. Chaudhary and S. Shukla, Sci.
Rep. 6, 31073 (2016).
[12] P. Hohenberg and W. Kohn, Phys. Rev. 136, B864 (1964).
[13] W. Kohn and L. J. Sham, Phys. Rev. 140, A1133 (1965).
[14] P. E. Blöchl, Phys. Rev. B 50, 17953 (1994).
[15] G. Kresse and G. J. Furthmuller, J. Phys. Rev. B 54, 11169 (1996).
[16] A. Togo and I. Tanaka, Scr. Mater. 108, 1 (2015).
[17] H. K. Monkhorst and J. D. Pack, Phys. Rev. B. 13, 5188 (1976).
[18] J. P. Perdew, K. Burke, M. Ernzerhof, Phys. Rev.
Lett. 77, 3865 (1996).
[19] V. I. Anisimov, J. Zaanen, O. K. Andersen, Phys.
Rev. B 44, 943 (1991).