항암제 개발: 토포아이소머라제, HSP90, mTOR 및 티로신키나제 억제제
최경철2, 이성호2, 권미지2, 홍세영2, 박희호1,2, 임광석1,2*
Development of Anticancer Chemotherapy:
Inhibitors of Topoisomerase, HSP90, mTOR and Tyrosine Kinase
Kyoungcheol Choi2, Seongho Lee2, Miji Kwon2, Seyoung Hong2, Hee Ho Park1,2, and Kwang Suk Lim1,2*
Received: 2 February 2021 / Revised: 29 March 2021 / Accepted: 29 April 2021
© 2021 The Korean Society for Biotechnology and Bioengineering
Abstract: Chemical anticancer drugs that have been used for a long time for the treatment of cancer have high anticancer effects and many side effects. The side effects of anticancer drugs have also affected normal cells because they induce the death of cancer cells by inhibiting or blocking essential mecha- nisms for cell survival. Recently, inhibitory drugs that inhibit specific mechanisms of cancer cells are receiving a lot of attention as anticancer drugs. Inhibitory drugs have less effect on normal cells by inhibiting the activity of target proteins that are overexpressed in cancer cells. Representative anticancer inhibitors among many inhibitory drugs are mTOR inhibitors, topoisomerase inhibitors, heat shock protein 90 (HSP90) inhibi- tors and tyrosine kinase inhibitors. For each of these inhibitor family, new candidate inhibitor drugs are continuously being developed and clinical trials are underway. In this review, we will examine the drug mechanism of each inhibitor drug, and describe the approved drugs and drugs in clinical trials.
Keywords: inhibitors, heat shock protein 90, mTOR, topoisom- erase, tyrosine kinase
1. INTRODUCTION
많은 OECD(Organization for Economic Cooperation and
Development) 국가에서 고령화가 증가하면서 암, 심근경색 및 뇌출혈 같은 3대 질환에 노출되는 인구의 비중이 높아지 고 있다. 암 발병이 증가하고 있지만 암 환자의 생존율 역시 높아지고 있는데, 이는 암 치료법이 다양해지고 정밀해지면 서 나타난 결과이다 [1]. 암 치료는 외과적 수술 (surgery), 화 학요법 치료법 (chemotherapy), 및 방사선 치료법 (radiation therapy) 을 대표적으로 이용하여 진행되고 있으며, 각각의 기 술들의 발전에 따라 치료 효과 역시 향상되고 있다. 또한, 면 역 반응에 대한 이해가 높아지면서 면역관문억제제와 면역 항암세포치료제 등이 도입되면서 암 환자의 생존율이 크게 높아지고 있다. 화학요법 치료제는 오랜 시간 동안 항암치료 제 활용되었으며 최근에도 다양한 항암제들이 보고되고 있 다. 특히 많은 항암제들이 표적전달체의 적용 및 면역항암제 와의 병용 요법에 활용되면서 효과는 높아지고 부작용은 개 선되어 암을 치료하는 일차 의약품으로 더 높은 관심을 받고 있다 [2].
화학요법 치료제로 이용되는 항암제는 대표적으로 80여 종이 되며 암세포의 생명 활동에 중요한 대사과정을 저해하 여 암세포의 성장을 억제하고 사멸을 유도한다 [3]. 암세포 성장을 억제하는 항암제의 기전은 대표적으로 다음의 4가지 로 구분할 수 있다. 1) Akylating agents; 2) Antimetabolites; 3) nti- microtubule agents; 4) Topoisomerase inhibitor. 대표적인 4가 지 기전은 세포의 중심원리 (central dogma)를 억제하여 세포 의 성장을 억제하게 되며 이는 정상세포 및 암세포에서 동일 하게 나타나는 기전이다. 이에 항암제를 암세포에 정확하게 전달하지 못하면 정상세포의 사멸로 인해 부작용이 나타나 게 된다. 따라서 이 부작용을 해결하기 위해 항암제의 표적 전달체 개발이 많이 진행되고 있으며, 암세포에만 특이적으 로 발현하는 표적 단백질에 선택적으로 작용하는 억제제 약 물에 대한 개발이 증가하고 있다.
억제제 (또는 저해제) 약물들은 세포 내의 필수 단백질에
1강원대학교 문화예술공과대학 생물공학과
1Department of Biotechnology and Bioengineering, Kangwon National University, Chuncheon 24341, Korea
Tel: +82-33-250-6279, Fax: +82-33-243-6350 E-mail: kslim@kangwon.ac.kr
2강원대학교 문화예술공과대학 바이오헬스기기 융합기술 협동과정
2Interdisciplinary Program in Biohealth-machinery convergence engineering, Kangwon National University, Chuncheon 24341, Korea
Review Paper
결합하여 단백질의 기전을 방해하게 되고 이를 통해 정상적 인 세포의 활동을 억제하고 나아가 사멸로 유도하게 된다.
특히 억제제 약물들은 암세포에서 특이적으로 과발현되는 단백질을 표적으로 하여 정상세포에 미치는 영향을 낮춰 부 작용을 개선하고 있다. 이처럼 암세포에 선택적으로 작용하 기 때문에 다양한 억제제 약물들이 개발되고 있으며 기존 억 제제 약물들의 단점을 개선한 신규 억제제 약물 후보물질들 이 지속적으로 보고되고 있다. 또한 최근 항체-약물 접합체 중 Enhertu® (Trastuzumab+Deruxtecan)가 성공적으로 시장 에 진입하여 억제제 약물의 활용이 확대되고 있어 억제제 약 물에 대한 관심이 더욱 높아지고 있다 [4]. 암세포에 특이적 으로 작용하는 억제제 약물로 다음의 4가지 억제제 약물 계 열을 선별하였다. 1) topoisomerase (Top) inhibitor; 2) heat shock protein 90 (HSP90) inhibitor; 3) mammalian target of rapamycin (mTOR) inhibitor; 4) tyrosine kinase inhibitor (TKI).
선별된 4가지 억제제 약물들의 기전은 세포의 생존에 중요 한 역할을 하는 단백질들을 표적으로 하여 약효를 나타내게 된다 (Fig. 1). 각각의 억제제 약물들은 승인을 받은 약물, 임 상 시험 중인 약물, 및 새로운 후보 물질 개발 단계로 다양하 게 연구 중이다. 본 총설에서는 대표적인 4가지 억제제 약물 에 대한 기전을 살펴보고 개발된 항암제 및 개발 중인 신규 항암제들에 대해 논의하여, 향후 억제제 약물들의 개발 및 신규 항암제 개발 방향을 알아보고자 한다.
2. TOPOISOMERASE INHIBITOR
Topoisomerase (Top)는 DNA 전사 및 복제 과정에서 DNA 가 닥의 꼬임 현상을 이완시키는 역할을 한다. DNA는 supercoil
형태로 이중 가닥이 풀리면서 전사 및 복제가 진행되는데 나 선 간의 공유 결합으로 인한 DNA 가닥의 꼬임 현상이 나타 나면 정상적인 전사 및 복제가 어렵게 된다. 이때 꼬임 현상 을 해결에 중요한 역할을 하는 topoisomerase는 세포의 생존 에 중요한 역할을 하는 효소이다. 진핵생물, 고세균 및 진균에 존재하며, 인간 세포는 6개의 유전자가 암호화되어 있고 박테 리아는 4개의 유전자가 암호화되어 있다. Topoisomerase는 DNA gyrase가 발견되면서 coumarin(coumarin-3-carboxylic acid) 와 quinolone의 표적으로 밝혀졌다 [5,6]. 그 이후로 topoisomerase는 많은 항암 및 항균 약물의 표적으로 활용되 고 있다 [7-9].
2.1. Function of topoisomerase
Topoisomerase (Top)의 유형은 크게 type 1과 type 2 (Topoi- somerase 1, Topoisomerase 2)로 구분되며 DNA-단백질 결합 의 극성, 작용 기전, 및 구조에 따라 세부적으로 Top1A, Top1B, Top2A 로 분류된다 [10,11]. Top1A는 DNA에 결합하 기 위해 단일 가닥이 필요하여 이중 가닥 중 한 가닥을 절단 하고 활성화된 티로신 잔기를 5′-phosphoryl group에 공유 결 합으로 부착한 후에 반대쪽 가닥을 통과시키고 절단한 가닥 을 재조립하여 DNA topology를 변화시킨다. 반대로 Top1B 는 이중 가닥 중 한 가닥을 절단하고 활성화된 티로신 잔기 를 3′-phosphoryl group에 부착시킨 후에 DNA를 회전시켜 DNA supercoils을 풀어준다. Top2A는 이중 가닥의 두 가닥 (gate segment)을 절단하고 또 다른 이중 가닥 (transfer segment)을 통과시킨 후에 봉합시킨다. 이때 2개의 활성화된 티로신 잔기와 한 쌍의 5′-phosphoryl group을 phosphotyrosyl bonds로 결합시킨다. 이와 같이 topoisomerase A는 절단한 가 닥을 온전한 가닥이 통과하는 방법을 가지며, topoisomerase B는 절단한 가닥이 온전한 가닥을 중심으로 회전하는 방식 을 가진다 [11,12].
2.2. Mechanism action of topoisomerase inhibitor (Topi) Topoisomerase inhibitor (Topi)는 Top1 및 Top2를 표적으로 하여 DNA의 전사 및 복제 과정을 방해하는 억제제이다 [7,12]. Topi는 크게 4가지 기전을 통해 topoisomerase를 억제 하여 항암 효과를 나타낸다. 첫 번째는 topoisomerase의 활성 부위에 inhibitor가 결합하여 DNA 기질이 결합하는 것을 방 해하는 기질 경쟁 억제 식이지만 아직 주목할 만한 예시가 없 다. 두 번째는 ‘Topoisomerase poisons’을 형성하는 것으로 가 장 활발하게 연구가 되었다. Topi의 결합에 따라 Ternary protein-DNA-drug complex 로 구성되며 DNA의 재결합을 방 지하고 효소를 절단 부위에 고정해 효소의 전환을 막아 DNA 복제 억제, 이중 가닥 절단 및 후속 세포 사멸 등의 독 성을 주게 된다. 세 번째는 촉매 억제제를 통해 효소 활성을 막는 것이다. 이는 Top1B와 Top2A 대상으로 한 촉매 억제제 로써 임상 단계에서 큰 주목을 받지 못했다. 하지만 암세포 에서 다량의 topoisomerase를 억제할 경우 촉매 억제제는 강 력한 항암제가 될 수 있을 것으로 보인다. 마지막으로 ATP Fig. 1. Mechanism of topoisomerase, HSP90, mTOR and tyrosine
kinase.
결합 부위를 억제하는 것으로, 이것은 Top2에서 나타나는 ATP 결합 부위에 Topi가 결합하여 Top2를 불활성화시킨다.
다양한 기전에 따라 Topi가 topoisomerase를 억제할 수 있는 데 그중 가장 많이 연구되고 있는 Top1B와 Top2A와 관련된 Topi를 살펴보고자 한다.
2.2.1. Topoisomerase inhibitor type 1 inhibitors
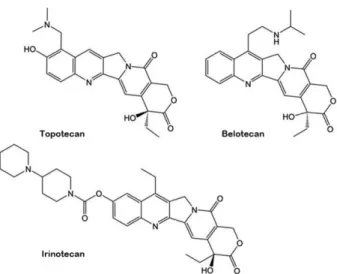
Human topoisomerase type1 (hTop1) inhibitor로는 quinoline alkaloids, camptothecin (CPT), topotecan (Hycamtin), 및 irino- tecan 등이 있다 [13]. 약물들은 Top 1-DNA 공유결합 복합체 에 결합하여 Top1을 공유결합 세포독성 부가 단백질로 변환 시킨다 [7,14]. 이것은 DNA 말단 사이에 약물 분자가 삽입된 모습을 보이며, DNA 복제 및 생성을 억제하고 이중 가닥 절 단을 발생시킨다. hTop1 poisons 형성 항암제로 현재 임상 연 구 중인 indolocarbazoles과 indenoisoquinolines가 있다 [15,18].
이것은 평면 다환식 중심체 형태를 가지며 camptothecin과 유사하게 가닥 절단 부위에 삽입되는 결합 방식을 가지고 있 다 [16,17,19,20]. 이들과 같이 대부분의 hTop1 inhibitor는 topoisomerase poisons을 형성하는 기전이 임상적으로 가장 효과적임을 보여주고 있으며, 촉매 억제 기전을 가진 inhibitor는 연구가 더 필요한 상황이다.
2.2.2. Topoisomerase inhibitor type 2 inhibitors
Human topoisomerase type 2 (hTop2) inhibitor 의 대표적인 기 전으로 topoisomerase poisons 형성과 hTop2 촉매적 억제가 있다. Topoisomerase poisons 형성은 human Topoisomerase 1 inhibitor 와 유사하게 끊어진 DNA 가닥에 결합하여 DNA의 복원을 방해한다 [21]. 그 예시로는 anthracyclines과 epipodo- phyllotoxins가 있으며 고형 종양 및 혈액학적 악성 종양의 치 료제로 사용 중이다. 그러나 이들은 심장 독성과 백혈병 유 발이라는 두 가지 심각한 부작용을 가지고 있다 [22, 23]. 심 장 독성은 anthracyclines의 reactive oxygen species (ROS)의 증가와 지질의 과산화로 인한 것이다. 그리고 백혈병 유발은 치료 관련 급성 골수성 백혈병 (t-AML)과 치료 관련 급성 프 로밀로세포 백혈병 (t-APL)가 대표적이며, t-AML은 mixed lineage leukemia (MLL) 유전자의 재배치에 의한 것이고, t- APL 은 promyelocytic leukemia (PML) 유전자의 전좌로 인해 서 발생한다. 현재는 부작용을 예방하기 위해 누적 투여량을 제한하고 있지만, 그럼에도 가장 널리 사용 중인 항암제 중 하나이다 [7].
2.3. Topoisomerase inhibitors family
현재 임상적으로 판매된 topoisomerase type 1 inhibitors로는
대표적으로 camptothecin 유도체가 있다 (Table 1, Fig. 2).
Camptothecin은 중국의 camptotheca acuminata의 껍질과 줄 기에서 분리되어 항암제로 사용되었다. 이것은 주로 topoiso- merase type 1B 대상으로 독을 형성시켜서 DNA의 복제를 막 고 종양 세포를 사멸시킨다. 그러나 낮은 용해도와 심한 부 작용으로 인해 사용이 중지되었다. 그렇기에 이에 파생된 topotecan (Hycamtin), irinotecan, belotecan (Camtobell inj.)가 개발되었다. Topotecan은 최초로 구두 투여를 위해 승인된 hTop1 inhibitor 로 난소암, 소세포 폐암 치료제로 사용되고, irinotecan은 결장암과 소세포 폐암을 치료하는 데 사용된다 [24,25]. Belotecan은 이 중 가장 새로운 camptothecin 유도체 로 2003년에 한국에서 승인되어 판매되고 있으며, 독성이 감 소한 효능을 가지고 있다. 그렇지만 E-ring으로 인한 화학적 불안정하며 골수 억제로 인한 부작용들 때문에 복용량에 제 한을 두고 있다 [26-28].
Topoisomerase type 2 inhibitors는 대부분 hTop IIA poison 을 이용한 제제이다 (Table 2, Fig. 3). 대표적으로 anthracycline 제제와 epipodophyllotoxin 제제가 있다. 임상적으로 판매된 anthracycline 제제로는 doxorubicin, epirubicin, valrubicin, daunorubicin이 있다. Doxorubicin은 유방암, 백혈병, 림프종, 육종, 암종 등의 기타 종양의 치료에 사용이 되고 있다. 하지 만 이들은 anthracycline가 세미퀴논 라디칼을 형성시키고, 이것이 활성산소종 (ROS)로 인한 산화 스트레스를 발생시 킨다. 그렇기 때문에 심장 독성을 발생시키는 부작용을 가지 고 있다 [29-31]. Epipodophyllotoxin은 미국의 mayapple plant (Podophyllum peltatum)에서 유래된 천연 물질이다 [32].
Epipodophyllotoxin 제제도 topoisomerase poison 기전을 이
Table 1. Summary of approved topoisomerase type 1 inhibitor family
Drug Target Target diseases Approval date Company
Topotecan Type IB poison Ovarian cancer 1996 GlaxoSmithKline
Irinotecan Type IB poison Colon cancer 1996 Pfizer
Belotecan Type IB poison Small cell lung cancer, Ovarian cancer 2003 Chong Kun Dang
Fig. 2. Structure of topoisomerase type 1 inhibitors [8].
용한 항암제이다. 판매된 약품으로는 etoposide와 teniposide 가 있다. 이것들은 소세포 폐암, 림프종 백혈병, 고환암, 방광 암, 융모성 질환 등을 치료하는 데 사용된다. 또한 기존 ATP 와의 결합에 의한 약효 감소를 피하기 위해 새롭게 보고 된 신규 화합물이 보고되고 있다 [33].
2.4. Development of topoisomerase inhibitors
FDA 승인을 받은 억제제들의 부작용 개선 및 효능 향상을 위 한 새로운 topoisomerase inhibitor들이 보고되고 있으며 임상 시험을 진행하고 있다 (Table 3). 임상 1상(Phase 1)을 완료한 물질 중 indenoisoquinolines는 National Cancer Institute (NCI) 에서 진행한 연구로 camptothecins에 비해 합성 및 화학적으 로 안정적이다. 그리고 ATP-binding cassette transporters
(ABC transporters), ATP-binding cassette super family G member 2 (ABCG2)와 P-glycoprotein (MDR1)를 발현하는 세포에서 주로 작용하며 장점이 있다. 현재 임상 2단계를 진행 중인 물 질로 AR-67(7-t-butyldimethylsilyl-10-hydroxycamptothecin)가 있으며, 친유성이고 지질 이중 층으로 분할되어 있어서 가수 분해로부터 안정적이다. 약물이 가진 10-hydroxy의 기능으 로 카복실레이트 약물의 높은 친화적 상호작용을 약화시켰 으며 AR-67은 임상 1단계에서 85% 이상의 락톤 안정성을 보여주었다.
마지막으로 임상 3단계 연구 중인 제제로는 pixantrone와 aldoxorubicin이 있다. 먼저 pixantrone은 CTI BioPharma에서 연구 중이며 이것은 효능을 저하시키지 않으면서 심장 독성 을 최소화하도록 만들어진 새로운 anthracenedione이다 [34].
단독 요법으로 European Medicines Agency에서 승인을 받았으 며 rituximab이 처방된 환자에서의 pixantrone의 역할을 확인하 기 위해 진행한다. 그리고 CytRx에서 개발 중인 aldoxorubicin 은 종양 표적 doxorubicin으로써 연조직 육종을 대상으로 하 는 제제로 EMCH(N-ε-maleimidocaproic acid hydrazide) 링커를 통해서 내인성 순환 알부민에 결합하여 종양에 우선으로 축적 된다. 이후 종양 환경에서 링커가 분열되며 doxorubicin이 방 출되는 기전을 가지고 있기 때문에 전신 독성을 감소시키고 치료 효능을 증가시킨다.
3. HEAT SHOCK PROTEIN 90(HSP90) INHIBITOR
HSP90(heat shock protein 90) 은 진핵 세포 내에 풍부한 분자 샤페론 (molecular chaperones) 중 하나로, 대부분의 조직에 서 전체 단백질의 약 1~2%를 구성한다 [35,36]. HSP90은 다 Table 2. Summary of approved topoisomerase type 2 inhibitor family
Drug Target Target diseases Approval date Company
Doxorubicin Top IIA Acute Lymphoblastic Leukaemias, Acute Myeloblastic Leukemia 1974 Carlo Erba Farmitalia Spa Daunorubicin Top IIA Acute Lymphocytic Leukemia, Acute Myeloid Meukemia 1979 Sanofi-Aventis
Etoposide Top IIA Kaposi’s sarcoma, Ewing's sarcoma, Lung cancer 1983 Novartis
Amsacrine Top IIA Leukemia, Refractory Leukemia 1983 Erfa Canada
Eptoposide Top IIA Testicular cancer, Lung cancer, Lymphoma 1983 E.R. Squibb & Sons, L.L.C.
Mitoxantrone Top IIA Acute Myeloid Meukemia 1987 Emd Serono
Idarubicin Top IIA Acute Lymphoblastic Leukemia, Chronic Myelogenous Leukemia 1990 Pharmacia and Upjohn Company LLC Teniposide Top IIA Acute Lymphocytic Leukemia, Hodgkin's Lymphoma 1992 E.R. Squibb & Sons, L.L.C.
Valrubicin Top IIA Bladder cancer 1998 Endo
Moxifloxacin Top IIA Acute Exacerbation of Chronic Bronchitis (AECB), Bacterial
Conjunctivitis, Skin Infections 1999 Stat Rx
Epirubicin Top IIA Non-Small Cell Lung Carcinoma (NSCLC) 1999 Pharmacia and Upjohn
Company L.L.C.
Dexrazoxane Top IIA Cardiomyopathy 2007 TopoTarget
Pixantrone Top IIA Non-Hodgkin's Lymphoma, Refractory Non-Hodgkin's lymphoma 2012 Cti Life Sciences Limited
Fig. 3. Structure of topoisomerase type 2 inhibitors [8].
양한 클라이언트 단백질들 (client proteins)의 안정성과 활성 을 유지하기 위해 필요하며, 클라이언트 단백질들은 세포 내 에서 신호 전달, 증식, 및 생존에 중요한 역할을 한다. 지난 몇 년 동안 300여 개 이상의 단백질이 HSP90의 클라이언트 단백질 로 밝혀졌으며, 막전위 타이로신 키나아제 (transmembrane tyrosine kinases; HER2, EGFR), metastable signaling proteins (Akt, Raf-1 and IKK), 돌연변이 된 신호 전달 단백질 (mutated signaling proteins; p53, v-Src), 키메릭 신호 전달 단백질 (chimeric signaling proteins; Bcr-Abl), 세포 주기 조절기 (cell cycle regulators; Cdk4, Cdk6), 및 스테로이드 수용체 (androgen, estrogen, progesterone receptors) 등이 포함된다 [36]. 이러한 클라이언트 단백질의 대부분이 암에서 변이되거나 과발현 되는 것으로 알려져 있다.
HSP90은 종양 형성과 악성으로의 진행에 중요한 클라이언 트 단백질의 성숙과 안정화에 관여하기 때문에 암세포는 특
히 HSP90의 기능에 의존하게 된다. 또한 종양에서의 저산소 증, 낮은 pH, 및 낮은 영양공급과 같은 스트레스 환경 조건은 단백질을 불안정하게 하므로 암세포는 HSP90의 발현이 높 게 나타난다. 이러한 HSP90에 대한 종양 세포의 비정상적인 의존도는 정상세포와 대조적으로 총 단백질의 약 4~6%를 구성한다.
3.1. Function of HSP90
HSP90은 세포 형질전환과 종양 진행을 유발하는 영양소, 산 소 결핍 및 독성물질에 대한 노출 같은 종양 미세환경의 변 화에 반응하여 당분해 대사와 산화적 인산화 사이의 평형을 조절하는 필수적인 역할을 한다 (Fig. 4) [35,36]. HSP90 샤페 론은 복잡한 대사 경로에 관여하는 단백질의 발현을 제어하 는 HSP90 의존성 신호 전달 경로를 변경함으로써 간접적으 로 대사 변화를 조절할 수 있다. 또한 일부 대사 효소의 안정 Table 3. Topoisomerase inhibitor family in clinical trial
Drug Phase NCT# Target diseases Current state Sponsor
Dibenzonaphthyridinones I 00942799 Solid Tumors Completed Genzyme, a Sanofi Company
Namitecan I 01748019 Solid Tumors Completed sigma-tau i.f.r. S.p.A.
Indenoisoquinolines I 01051635 Neoplasms, Lymphoma Completed National Cancer Institute (NCI)
AR-67 I 01202370 Solid Malignancies Completed Arno Therapeutics
Vosaroxin II 02658487
Acute Myeloid Leukemia, Acute Myeloid Leukemia Arising From Previous Myelodysplastic Syndrome, ETC
Active, not recruiting
Vanderbilt-Ingram Cancer Center
CRLX101 II 01380769 Non-Small Cell Lung Cancer Completed NewLink Genetics
Corporation Pixantrone + Rituximab,
Gemcitabine + Rituximab III 01321541
Diffuse Large B-cell Lymphoma, de Novo DLBCL, DLBCL Transformed From Indolent
Lymphoma, Follicular Grade 3 Lymphoma
Completed CTI BioPharma
Aldoxorubicin III 02049905 Metastatic, Locally Advanced or Unresectable
Soft Tissue Sarcoma Completed CytRx
Fig. 4. Overview of HSP90 function in the cancer cells and normal cell (a) and HSP90-interacting co-chaperones and HSP90-dependent
client proteins (b) [32,35].
성과 형태, 기능적 활성을 제어함으로써 직접적으로 대사를 조절할 수 있다.
이러한 HSP90은 암세포에서 대사 핵심 규제에 관련된 여러 세 포 신호 경로를 상호작용하고 변형시킨다 [37]. HSP90은 글루 코오즈 트랜스포터 (Glucose transporter, GLUTs) 글루타민 트랜 스포터 (SLC2A5), 젖산염 탈수소효소 (lactate dehydrogenase, LDHA), 및 피루베이트 키나아제 근육 이소자임2 (Pyruvate kinase muscle isozyme, PKM2)의 발현을 조절하는 c-MYC의 접힘, 안정성, 및 활성을 조절한다 [38]. 이는 생합성 경로에 사용되는 당분해 중간체의 과잉 생산으로 인해 당분해 속도 증가를 유발한다. 저산소 상태에서 HSP90은 PFK1 (phospho- fructokinase 1), GAPDH (glyceraldehyde 3-phosphate dehydro- genase), LDHA(lactate dehydrogenase A), 및 HK II (hexokinase II)와 같은 GLUTs와 해당효소를 암호화하는 유전자의 발현 을 유도하는 HIFα (hydroxia-inducible factor 1 alpha)를 안정 화시킨다. 또한 HSP90의 억제는 HSP90 클라이언트 단백질 복합체를 변화 시켜 활성 감소를 일으키며, 그 후 클라이언트 단백질의 유비퀴틴 매개 단백질 분해와 동시에 oncoprotein 의 고갈, 악성 표현형의 종양 형성 신호 전달 경로를 통해 신 호를 하향 조절한다 [36].
3.2. Mechanism action of HSP90 inhibitor
대부분의 HSP90 억제제는 N-말단의 ATP 결합 포켓 (ATP binding pocket)을 차단하고 co-chaperone과 client protein 로 딩에 필수적으로 필요한 구조적 변화를 억제한다 (Fig. 5(a)) [39-41]. Geldanamycin (GA)는 HSP90의 N-말단 ATP 결합 포 켓에 결합하여 HSP90의 구조 변화를 방해하여 기능을 저해 시킨다 [42]. GA는 ATP와 비교하여 ATP 결합 부위에 대해
~100배 더 높은 친화성을 갖는다. GA는 HSP90에서 Leu48, Asn51, Gly97, Thr184, 및 Gly137에 수소결합을 형성하고, Asn55, Ile96, Met98, Asp102, Asn106, Leu107, Phe138, 및 Val150 에 소수성 결합을 형성한다 (Fig. 5(b)) [43]. GA와 그 유사체들은 HSP90을 ‘닫힌(Closed)’ 형태로 유지하고 ATP
주기의 촉진을 방지한다. HSP90에 의해 통제받지 못한 client protein은 proteasomic machinery에 의해 분해 된다.
HSP90의 또 다른 억제제인 Radicicol은 p60 v-src 단백질 키 나아제의 고갈과 Kras 활성 신호 전달 경로의 억제를 통해 in vitro와 in vivo에서 암세포 증식 및 혈관 신생을 억제할 수 있 다. 또한 HSP90의 N` 말단 도메인에 경쟁적으로 결합하여 HSP90 복합체 형성을 방해한다 [44].
3.3. HSP90 inhibitors
3.3.1. Geldanamycin (GA) derivatives
Geldanamycin (GA)는 1970년 Streptomyces hygroscopicus 의 발효 배양액에서 처음 분리된 벤조퀴아닌 안사마이신 (benzoquinone ansamycin)이다 (Fig. 6) [45,46]. GA는 처음 src 키나아제의 직접적인 억제제로 구분되었지만, HSP90에 직접적으로 결합하여 HSP90-v-src 이종 복합체의 형성을 방 해하는 것으로 나타났다. 또한 GA는 N-말단 도메인의 뉴클 레오타이드 결합 부분에 결합하기 위해 ATP와 경쟁함으로 써 HSP90의 ATPase 활성을 억제하여 클라이언트 단백질의 유비퀴틴 매개 프로테아좀 분해를 일으킨다 [47,48]. GA는 in vivo 실험에서 강력한 항암효과가 나타났지만, 용해도와 생체 내 안정성이 제한적이며 동물 모델에서 간독성이 높게 관찰되었기에 임상적 이용가능성이 없는 것으로 판명되었 다. 이에 GA과 유사한 항암 효과를 가지지만 독성이 낮은 GA derivatives에 대한 연구가 진행되었으며 대표적인 GA derivatives 에는 17-AAG(17-Allyl-17-Demethoxygeldanamycin), 17-DMAG(17-desmethoxy-17-N, N-dimethylaminoethylaminogel danamycin), IPI-504 등이 있다 [49,50].
17-AAG는 GA와 유사하지만, 독성 프로필이 개선된 화합 물이다. 또한 17-AAG는 전임상 모델에서 높은 항암 활성과 생물학적 활성이 예측되었지만, in vitro에서 세포성색전 (cytostasis)을 일으키거나 세포자멸사를 유발할 수 있다. 17- DMAG 는 GA의 C-17 메톡시 그룹을 N, N-dimethylethylamine 로 대체하여 만들어졌다. 17-DMAG는 이온화 가능한 아미
Fig. 5. The HSP90 chaperone cycle (a) and HSP90 biding site with HSP90 inhibitors (b) [32,36]
노 그룹이 존재하기 때문에 17-AAG에 비해 수용성이 증가하였 으며, 경구 생체 이용률과 더 큰 항종양 효과를 나타냈다 [50].
IPI-504 (17-allylamino-17-demethoxygeldanamycin hydroquinone hydrochloride)는 17-AAG의 또 다른 수용성 하이드로퀴논 하이드로클로라이드 유사체이다. IPI-504는 디티온산나트륨 (sodium dithionite)으로 17-AAG를 환원시킨 다음, 17-AAG 의 염산염으로 전환하여 제조할 수 있다. IPI-504와 17-AAG 는 실제로 산화환원효소의 작용을 통해 하이드로퀴논 (IPI- 504와 유사)과 퀴논 (17-AAG와 유사)의 형태 사이에 in vivo 에서 산화환원 평형으로 존재한다. 하이드로퀴논 형태인 IPI-504는 HSP90의 강력한 억제제이다.
3.3.2. Purine Derivatives
HSP90은 다른 ATP/ADP 결합 단백질에 대한 저분자에 의한 선택적 억제를 가능하게 하는 결합 뉴클레오타이드 (bound nucleotide)에 의해 채택된 독특한 형태를 가지는 대부분의 다른 ATPas들 사이에서 독특한 접힘을 가진다 [51]. 이러한 독특한 접힘의 이점을 이용하고 구조 기반 접근 방식을 사용 하여, 최초로 합성 HSP90 억제제로 만들어진 것이 퓨린-스 캐폴드를 기반으로 만들어진 PU3이다. PU3는 퓨린-스캐폴 드 클래스의 프로토타입으로서, 의약품 특성이 개선된 강력 한 선택적 억제제를 토출하기 위한 다양한 전략을 통해 최적 화되었다. 임상시험이 진행된 대상에는 퓨린 CNF2024/BIIB021, MPC-3100, PU-H71뿐만 아니라 퓨린과 유사한 Debio 0932 (CUDC-305)가 포함된다 (Fig. 6).
3.3.3. Resorcinol Derivatives (RD)
레조르시놀 유사체 (Resorcinol Derivatives, RD)는 1953년에 처음으로 곰팡이 Monosporium bonorden으로부터 분리된 마 이크로사이클릭 락톤 (macrocyclic lactone) 항생제다 [52].
RD 는 혈청에서 안정하지 않고 in vivo 활성이 낮지만, STA- 9090뿐만 아니라 NVP-AUY922, KW-2478, AT13387 등이 임상시험 진행 중이다 (Fig. 6). 이러한 분자들은 RD의 직접 적인 변형을 통해 발견되진 않았지만, 레조르시놀 코어를 결 합의 중요 요소로 유지함으로써 유사하다.
3.4. Development of HSP90 inhibitor 3.4.1. Geldanamycin derivative
HSP90 억제제에 대한 임상 진행은 geldanaymycin derivatives, purine derivatives, resorcinol derivatives 계열 약물들 모두 활 발히 진행되고 있다 (Table 4). Tanespimycin(17-AAG)은 HSP90 inhibitor 중 가장 처음으로 임상시험에 들어간 약물이다 [50].
Phase I 임상 단계에서 설사, 메스꺼움, 구토, transaminase elevation 과 피로를 포함한 독성이 나타났다. 또한 전이성 흑 색종 (metastatic melanoma) 환자를 대상으로 한 후속 phase II 임상 시험에서 MARK 경로의 지속적인 억제는 치료 후 생 체검사에서 관찰되지 않았다. 17-AAG는 계란 인지질과 4%
DMSO를 포함하는 희석제를 필요로 하므로 추가적인 독성
을 유발하게 된다. 따라서 실제로 이 화합물에 대한 소아
phase I 임상 단계에서 DMSO의 부피는 용량증대 (dose
escalation)의 제한 요소가 되었다. 유방암과 다발성 골수종
Fig. 6. Structure of heat shock protein 90 (HSP90) inhibitors [38].
에서 작용제를 통합한 결합요법의 유망한 활성과 제형의 개 선에도 불구하고, 17-AAG의 개발은 소유주인 Bristol-Myers Squibb에 의해 중단되었다. 17-DMAG는 17-AAG과 대비하 여 수용성이 개선되었으며, 여러 용량, 투여 일정, 및 투여 방 법으로 테스트되었다 [49]. 이러한 17-DMAG는 전립선암과 흑색종에서 항암 효과가 관찰되었지만 폐렴, transaminitis, 및 시력 변화를 포함한 독성에 의해 개발이 제한되었으며, 17-DMAG의 개발은 용량, 투여 빈도, 표적 억제 기간 및 독 성을 조절하는 어려움으로 제한적으로 진행되고 있다.
IPI-504 는 phase II/III 임상시험이 진행되었으나 17-AAG와 17-DMAG와 마찬가지로 주요 용량 제한 독성 (dose-limiting toxicity)은 간 독성이었다 [53]. 위장 기질 종양 (gastrointestinal stromal tumors, GIST) 환자를 대상으로 한 phase III 임상시 험 결과에서 4명의 사망자가 발생하여 연구가 조기 종료되 었다. 대조적으로 pre-treated 된 NSCLC 환자를 대상으로 동
일한 복용량과 투여 일정으로 진행한 phase II 임상 시험에서 는 허용 가능한 안전 프로파일이 입증되었는데 IPI-504의 독 성 프로파일은 높은 용량과 투여 일정에 따라 나타난다.
3.4.2. Purine Derivatives
BIIB021는 phase I/II 임상시험에서 만성 림프구성 백혈병 환 자 1명은 림프절 크기가 39% 감소하였으며, 16명을 대상으 로 한 평가 중 11명이 안정 (stable disease, SD)을 나타냈다 [54,55]. 이러한 데이터를 바탕으로 아로마신 (Aromasin)과 결합하여 phase II 임상 시험이 계획되었으나 BIB021가 개발 사인 Biogen은 진행하지 않았다. MPC-3100은 phase I 임상 시험을 완료하였나, 용해성과 생물학적 이용 가능성이 낮기 때문에 전구약물 (pro-drug)인 MPC-0767 개발 계획 중이다.
PU-H71은 진행성 고형암, 림프종, 골수종 장애를 가진 환자 를 대상으로 phase I 임상 시험에서 평가되고 있다.
Table 4. Hsp90 inhibitor family in clinical trial
Class Drug Phase NCT# Target diseases Current state Sponsor
Geldanamycin Derivatives
Tanespimycin (17-AAG, KOS-953)
II 00118248 Advanced or metastatic thyroid cancer Completed National Cancer Institute (NCI)
III 00546780 Multiple myeloma Completed Bristol-Myers Squibb
Alvespimycin
(17-DMAG) I 00803556 Advanced Solid Tumor Malignancies,
Breast cancer Completed Bristol-Myers Squibb Retaspimycin
(IPI-504) II 01362400 NSCLC (Non Small Cell Lung Cancer) Completed Infinity Pharmaceuticals, Inc.
Purine Derivatives
CNF2024/
BIIB021 I 00344786 B-Cell Chronic Lymphocytic Leukemia Terminated Biogen
MPC-3100 I 00920205 Cancer Completed Myrexis Inc.
PU-H71 I 01393509 Metastatic Solid Tumor, Lymphoma, Myeloproliferative Neoplasms (MPN)
Active, not recruiting
Memorial Sloan Kettering Cancer Center Debio-0932
(CUDC305) I 01168752 Advanced solid tumors, Lymphoma Completed Debiopharm International SA
Rasorcinol Derivatives
Ganetespib (STA-9090) II
01273896 Breast cancer Completed
Memorial Sloan Kettering Cancer Center/ Synta Pharmaceuticals Corp.
01031225 01167114 01200238 01173523 01039519
NSCLC Esophagogastric cancer
Oclular melanoma SCLC (Small Cell Lung Cancer) GIST (Gastrointestinal Stromal Tumor)
Completed Synta Pharmaceuticals Corp.
NVP-AUY922 (VER52269)
I/II 00526045 Breast cancer, Hematologic neoplasms Completed
Novartis Pharmaceuticals
II 01124864 NSCLC Completed
KW-2478 I 00457782
Multiple myeloma, Chronic Lymphocytic Leukaemia (CLL), B-cell Non-Hodgkin's
Lymphoma
Completed Kyowa Kirin Pharmaceutical Development, Inc.
I/II 01063907 Multiple myeloma, Completed
AT13387 I
00878423 Metastatic solid tumors Completed Astex Pharmaceuticals, Inc.
01246102 Solid tumors, Breast cancer Completed National Cancer Institute (NCI)
3.4.3. Resorcinol Derivatives
Ganetespib (STA-9090)은 단일 제제 임상 활성이 비소세포폐 암 (Non-small-cell lung carcinoma, NSCLC), 유방암, 위암, 흑 색종 및 장암에서 확인되었다 [56]. 가장 빈번한 부작용은 일 시적인 설사인데, 운동성 감소제로 관리가 가능했다. 단일 제제로 유방암에 대한 phase II 임상 시험의 초기 결과로, 진 행된 질병에 대해 이전에 최대 3개의 화학요법을 받은 환자 에게서 9%의 객관적인 반응률이 나타났다. 삼중 음성 유방 암 (triple-negative breast cancer)으로 치료받은 3명 중 1명은 경미한 반응/안정된 질병이 나타났으며, 이러한 결과는 HSP90 의 억제 효과가 phospho-STAT3+삼중음성유방암 세포에 대 해 매우 효과적인 것으로 밝혀진 전임상 연구와 일치한다.
4. MTOR(MAMMALIAN TARGET OF REPAPMYCIN) INHIBITOR
Rapamycin의 발견은 1965년 이스터 섬의 Rapa Nui에서 자 연적으로 발생하여 발견된 Streptomyces hygroscopicus (S.hygroscopicus) 균주에서 분리 배양을 바탕으로 1970년에 S.hygroscopicus의 Secondary metabolite 중 하나로 항진균 치료제로써 연구가 진행되었다 [57]. 또한 rapamycin의 이 용은 항진균제로의 응용뿐만 아니라 동물세포 내에서의 성 장 및 대사작용에도 영향을 끼쳐 종양에 치료제로서의 가 능성과 면역 억제제로서의 작용이 대두되었다 [58,59]. 이 러한 Rapamycin은 동물세포 내의 특이적인 단백질들을 목 표로 작용하게 되며, 세포의 성장과 대사과정을 억제하게 되
는데, 이러한 rapamycin의 기전에 대한 연구자들의 흥미는 rapamycin의 목표 단백질인 mTOR1과 mTOR2 복합체에 대 한 발견으로 이루어지게 된다 [60].
mTOR 는 세포 내에서 생장과 대사작용을 조절하는 역할을 해주는 중요한 단백질 kinase이다 (Fig. 7). mTOR는 세포 내 에서 mTOR complex (mTORC/mTOR 복합체)를 구성하는 촉매 서브 유닛으로 mTORC는 크게 두 가지로 구분된다.
mTORC1은 mTOR의 조절 단백질 (Raptor), mLST8/GβL, Deptor 및 프롤린이 풍부한 Akt 기질 40으로 구성된다.
mTORC2는 mTOR, Rictor (rapamycin 비민감성 동반자), mLST8/ GβL, Protor, Deptor 및 포유류 스트레스 활성화 단백 질 키나아제 상호 작용 단백질 (mSIN1)으로 구성된다 [61].
mTOR1은 영양소, 성장 인자, 에너지 신호 및 세포 스트레스 를 포함한 다양한 환경 신호에 의해 조절된다. mTORC2의 기능은 mTORC1에 비해 잘 알려져 있지 않지만 성장 인자에 의해 직접적 또는 간접적으로 자극되는 것으로 알려져 있다 [62]. 이러한 mTORC 작용은 세포사멸, 성장, 자식작용의 영 향을 미쳐 세포의 생장을 조절하므로 골다공증, 당뇨병 등과 같은 다양한 만성 질환의 발병에 영향을 끼치며, 대표적으로 암 발병에도 연관이 있음이 시사되었다 [63-65].
4.1. Function of mTOR
mTOR는 세포 내의 대사 과정 중 중간 물질로서의 역할이 되 는 경우가 많이 존재한다. 다양한 요인들에 의한 mTOR의 활 성화는 당, 아미노산, WNT signaling, 성장인자와 같은 외부 의 물질이 세포 내의 신호 전달을 전달시키면서 발생하는 물 질에 의해 진행되게 된다. mTOR의 활성화는 다양한 세포의
Fig. 7. mTOR signal pathway in the cell [54].
생명활동으로 이어지는 만큼 배아 줄기세포에서도 중요한 역 할을 수행하는데, 이는 쥐의 배아줄기세포 (ESC; Embryonic stem cell)에서의 mTOR 유전자 knock out을 통해 mTOR 유 전자의 음성 세포를 구축하였고, 이는 배아줄기세포의 생명 활동에 치명적임을 시사하였다 [66,67]. 또한, mTOR는 당, 단백질, 및 지방의 이화작용 및 전반적인 세포내 에너지 저 장을 관리하여 세포의 생장 및 성장을 조절하는 단백질이다.
이러한 mTOR의 작용은 에너지의 공급이 적어질 때, mTOR 의 작용이 정상적으로 저해되어 항상성을 유지하도록 조절 된다. 그러나 암세포 내에서는 이러한 mTOR활성화의 조절 이 제대로 이루어지지 않아 비정상적으로 mTOR관련 대사 과정이 활성화가 이루어지는 것이 관찰되었다 [68].
mTOR 단백질 관련 대사과정의 과활성화는 암세포의 발현 을 촉진시키는 것을 확인할 수 있었다. 일반적인 breast epithelial cell에 oncogenic PI3K (H1047R)와 K-Ras (G12V) 의 발현은 mTOR 단백질의 대사를 촉진하여, de novo lipogenesis를 촉진하여 유방암세포로의 분화를 유도하였다 [69]. 또한 방광암세포에서 Pyruvate kinase M2 (PKM2) 과발 현에 의해 Fatty acid synthase (FASN)의 종결 단계가 활성화 되어 방광암 세포 내의 lipogenesis가 과활성화되는 것과 더 나아가 AKT/mTOR signaling pathway를 저해해주는 sterol regulatory element binding protein 1c (SREBP 1c)의 발현이 저해되는 것을 통해 암세포 내의 mTOR의 작용이 과활성화 되는 것을 확인할 수 있었다 [70]. mTOR는 암의 진행 및 전 이에도 중요한 역할을 하는데 mTORC1과 mTORC2는 대장 암에서 상피-간엽 전이, 운동성 및 전이에 밀접한 것으로 나 타났다. mTORC2는 유방암 및 신경 교종과 같은 암에서 전 이에 중요한 역할을 하는 것으로 알려져 있다. 암세포 내에 서 mTOR 단백질의 대사촉진은 암세포의 생장 및 분화에 필 요한 영양분공급에 영향을 미쳐 암세포 생장을 활성화시키 는 작용한다.
4.2. Mechanism action of mTOR inhibitors
mTOR 복합체가 암세포의 성장 및 증식에 중요한 역할을 하 며 이 복합체 구성을 위해서는 mTOR가 촉매 역할을 해야 한
다. 복합체 형성 단백질 중 FKBP12 단백질은 mTOR의 FRB 부위에 결합하여 복합체 형성을 관여하는데 rapamycin은 이 단백질에 비가역적으로 결합하여 키나아제 활성을 억제한 다 (Fig. 8) [71]. Rapalogos는 rapamaycin과 유사한 작용 메커 니즘을 유지하여 1세대 약물인 Termsirolimus와 2세대 everolimus는 각각 FKBP12와 cyclophilin FKBP-1에 결합하 여 mTORC1의 활성을 저해한다. 3세대 저해제인 Halitulin analog ICSN3250은 mTOR 단백질내의 FRB domain 상에 존 재하는 phospholipids acid와 경쟁적으로 작용하여 억제하는 효과를 나타낸다. 또한 LY3023414약물의 경우 시험관 내 실 험에서 PI3K/mTOR/DNA 의존성 protein kinase (DNA-PK) pathway 를 저해 시켜 암세포의 성장을 억제하게 된다. mTORC1 에 대해서는 직접적으로 작용하는 반면 mTORC2는 일반적 으로 rapalogs에 반응이 낮고 간접적인 것으로 알려져 있다.
약물에 대한 장기 노출에 따라 mTORC2의 조립이 억제되고 이후 Akt 신호 전달이 억제되어 효과가 나타나게 되는데 급 성 골수성 백혈병 환자에서 termsirolimus와 everolimus의 치 료효과를 통해 기전이 입증되었다.
4.3. mTOR inhibitors
체내 암세포의 성장을 위해선 영양공급 및 비정상적인 분열 과정이 이루어지므로 대부분의 암세포에서는 mTOR 단백질 의 과활성화가 이루어지는 것을 발견했고, 이를 억제하기 위한 억제제 개발이 진행되었다. 1세대 약물인 temsirolimus (Torisel®)와 2세대 약물로는 everolimus (Afinitor®)를 이용 하여 신장 및 유방암 치료제로 FDA 승인이 진행되었다 [72,73]. Temsirolimus는 고도 renal cell calcinoma (RCC)치료 에 사용되었다. Everolimus는 exemestane (Aromasin®)과의 병용 요법을 통해 sunitinib (Sutent®) 또는 sorafenib (Nexavar®) 에 의한 치료가 불가능한 RCC치료제로 적용 중이며, 호르몬 수용체 양성 및 Her2 음성 유방암, pancreatic neuromedicinal tumors (pNET), subependymal giants-cell astrocytoma (SEGA)까 지 적용되어 temsirolimus보다 더 다양한 암종 치료에 응용되 고 있다.
Fig. 8. Structure and function of mTOR complex 1 and 2 [64].
4.4. Development of mTOR inhibitors
더 나아가 3세대 mTOR 억제제로서의 응용 및 연구가 진행 되고 있다 (Table 5). Rapalink-1은 mTOR 단백질에 대한 두 가지 target 유전자에 동시에 작용한다. Rapalink-1은 강력한 항암효과를 가지며, 기존의 1세대 및 2세대 mTOR 억제제의 내성을 가지는 종양의 치료가 가능하다. 이러한 rapalink-1의 제작 방법은 새로운 항암제, 항바이러스제, 항균제 등의 설 계를 위한 성공적인 모델로 보여진다 [74]. 또한 1세대 및 2 세대 mTOR 억제제가 아닌 새로운 물질을 이용한 3세대
mTOR억제제의 발견이 이루어지고 있다. Halitulin analog ICSN3250는 mTOR 단백질 내에 FRB domain 상에 존재하는 phospholipids acid와 경쟁적으로 작용하여 이를 대체하고, mTOR 의 저해를 일으키는 새로운 억제제로 보이고 있다 [75].
또한 이전에 발견된 LY3023414 약물의 경우 시험관 내 실험 에서 PI3K/mTOR/DNA 의존성 protein kinase (DNA-PK) pathway 를 저해 시켜 암세포의 생장을 막는 기전이 밝혀졌고, 임상실 험에서, phosphoinositide-3-kinase regulatory subunit 1 (PIK3R1) 및 PTEN mutations을 가지는 자궁내막 종양에서의 지속적 Table 5. mTOR inhibitor family in clinical trial
Drug Phase NCT# Target diseases Current State Sponsor
Temsirolimus I
01050985 Advanced Solid Tumors Completed Georgetown University
01065662 Endometrial Cancer, Ovarian Cancer, Cervical
Cancer, ETC Completed Susana M. Campos, MD
II
00838955 Hodgkin's Lymphoma Terminated Loyola University
01687673 Hepatocellular Carcinoma Completed University of California, San Francisco
III 04433572
Alveolar Rhabdomyosarcoma,Botryoid-Type Embryonal Rhabdomyosarcoma, Embryonal
Rhabdomyosarcoma
Recruiting National Cancer Institute (NCI)
IV 01180049 Non-Hodgkin's Lymphoma Completed Pfizer
Everolimus I
02138929 Esophageal Cancer Completed M.D. Anderson Cancer Center
01430572 Advanced Cancers, Solid Tumors Active,
Not recruiting M.D. Anderson Cancer Center
II
01661283 Malignant Peripheral Nerve Sheath Tumors,
MPNST, Sarcoma Completed Sarcoma Alliance for Research through Collaboration 01263951 Differentiated Thyroid Cancer Completed Abramson Cancer Center of the
University of Pennsylvania III 01524783 Advanced NET of GI Origin, Advanced NET
of Lung Origin Neuroendocrine Tumors Completed Novartis Pharmaceuticals IV 01514448 Metastatic Renal Cell Carcinoma (mRCC) Completed Novartis Pharmaceuticals
03525834 Renal Angiomyolipoma Completed Novartis Pharmaceuticals
Sunitinib I
03900793 Osteosarcoma Completed University of Colorado, Denver
02164240 Gastrointestinal Stromal Tumor Active,
Not recruiting Dana-Farber Cancer Institute
II 02713763 Pancreatic Neuroendocrine Tumour Metastatic Completed Grupo Espanol de Tumores Neuroendocrinos
03066427 Clear Cell Renal Carcinoma Completed NCT03066427
III
03673501
Clear Cell Renal Cell Carcinoma, Stage I Renal Cell Cancer AJCC v6 and v7, Stage II Renal Cell Cancer AJCC v7, Stage III Renal
Cell Cancer AJCC v7
Completed Deciphera Pharmaceuticals LLC
01164202 Liver Cancer Completed Federation Francophone de
Cancerologie Digestive IV 01525550 Well-differentiated Pancreatic Neuroendocrine
Tumor Completed Pfizer
LY3023414 II 02407054 Prostate Cancer Metastatic Completed Eli Lilly and Company
04032080 Triple Negative Breast Cancer Recruiting Baylor Research Institute
인 부분적 관해에 결과를 나타내었다 [76].
이러한 mTOR 억제제의 항암제로서의 다양한 응용은 연구 가 지속되고 있으며, 빠른 속도로 진행되고 있다. 또한 다양 한 약물들이 임상에서 시도되고 있으며, 더 나아가 새로운 항암제가 개발될 것으로 전망되고 있다.
5. TYROSINE KINASE INHIBITOR
Protein tyrosine kinase (PTK)는 ATP를 이용하여 단백질의 특 정 tyrosine residues를 인산화하고 이를 통해 세포의 proli- feration, differentiation, migration, metabolism 및 programmed cell death에 관여한다 [77]. PTK는 수용체 PTK (RTK) 및 비 수용체 PTK (NRTK)으로 나눌 수 있다. RTK에는 표피 성장 인자 수용체 (Epidermal growth factor receptor, EGFR), 혈소 판 유도 성장인자 수용체 (Platelet-derived growth factor receptor, PDGFR), 혈관 내피 성장인자 수용체 (Vascular endothelial growth factor receptor, VEGFR), 인슐린 수용체 (Insulin receptor, InsR) 계열 등이 있다 [78]. 그들은 보통 특정 ligand에 결합하 는 세포 외 region, transmembrane 영역, 및 substrate에 선택 적으로 결합하고 인산화하는 세포 내 kinase domain을 가지 고 있다. RTK는 표적 단백질의 ligand 및 인산화 된 tyrosine residues에 결합할 수 있으며 여러 signal pathway를 통해 signal 을 전달하여 일련의 생화학적 반응 활성화 및 종합적 인 세포 반응 (예: 세포 증식)을 일으킬 수 있다 [79]. 암에 대
한 임상 연구는 이러한 수용체와 그 ligand가 많은 종양에서 중요하다는 것을 보여주었고, 많은 암들은 과도한 tyrosine 인산화 signal을 세포로 유발하는 성장인자를 지나치게 많이 가지고 있다. NRTK는 일반적으로 세포 외 region이 없다. 이 들은 보통 세포막과 결합되거나 세포질 내에 존재하는데, Abl kinase, Src kinase 계열 등이 포함된다. NRTK는 주로 cytokine 수용체, T세포 수용체 및 기타 signal pathway를 통 해 signal 전달을 수행한다 [80]. T 림프구 수용체, B 림프구 수용체, 및 immunoglobulin 수용체 등은 NRTK를 모집한 후 tyrosine 인산화를 통해 signal 전달 복합체를 형성한 후 downstream signal 전달을 활성화하고 세포 증식을 촉진하며 종양 형성을 유도할 수 있다. 따라서 PTK의 작용을 억제하 는 tyrosine kinase inhibitor (TKI)에 대한 연구가 활발하게 진 행되고 있다.
5.1. Function of tyrosin kinase
수용체 tyrosine kinase의 세포 외 region에 성장인자를 포함 하는 신호 전달 단백질 및 호르몬이 결합되면 세포 내 region 에 ATP가 결합한다 (Fig. 9) [81-84]. 그 후 수용체는 ATP로부 터 인산기를 제거하고 이를 수용체 또는 effector 단백질 상 의 tyrosine residues로 옮겨 인산화한다. 인산화된 tyrosine residues 은 수용체에 연결된 effector 단백질의 docking site로 작용하고 PI3K/AKT/mTOR signal pathway, RAS/RAF/MEK/
ERK signal pathway, JAK/STAT signal pathway를 통해 세포 증식, 세포 생존, 단백질 합성 등에 관여하는 transcription
Fig. 9. Summary of signaling mechanism of receptor tyrosine kinase(RTK) [76].
factor (예를 들어 성장인자)를 발현시킨다. NRTK 또한 cytokine 수용체, T 림프구 수용체 등의 신호에 의해 활성화 되어 ATP의 인산기를 tyrosine residues로 옮겨 인산화시키고 RAS/RAF/MEK/ERK signal pathway 또는 STAT signal pathway를 통해 세포 증식을 촉진한다. TKI는 이러한 PTK의 pathway에서 RTK와 NRTK에 존재하는 ATP 또는 신호 전달 단백질 및 호르몬 결합 부위에 결합하거나 특정 kinase에 결 합하여 표적의 형태적 변화를 유도한다. 이를 이용해 tyrosine residues 인산화를 억제하여 tyrosine kinase의 활성을 저하시 킨다 [85].
5.2. Mechanism action of tyrosine kinase inhibitors 지금까지 알려진 대부분의 TKI는 ATP와 경쟁적으로 결합하 는 것으로 알려져 있다 [84,86]. ATP는 Tyrosine kinase에 adenine ting에서 tyrosine kinase의 ATP 결합 cleft까지 수소 결합이 형성되면서 결합하게 된다 [87]. TKI는 tyrosine kinase 의 ATP 결합 부위에 특이적으로 결합하여 ATP의 결합 을 방해함으로써 tyrosine kinase의 활성을 저해한다. I 형 TKI 는 tyrosine kinase의 활성 형태를 인식한다. I 형 TKI는 ATP 에 의해 일반적으로 형성되는 수소 결합을 모방하는 1~3개 의 수소 결합으로 ATP 결합 부위에 결합한다. I 형 TKI와 달
리, II 형 TKI는 tyrosine kinase의 비활성 형태를 인식한다. II 형 억제제는 ATP 결합 부위에 직접 인접한 소수성 pocket을 점유하여 ATP와 간접적으로 경쟁합니다. 이 소수성 pocket 은 activation loop의 DFG-out 형태에 의해 생성됩니다. 이 독 특한 DFG-out 형태는 allosteric site라고도하며, II 형 억제제 는 allosteric 방식으로 tyrosine kinase 활성을 조절할 수 있습 니다. III 형 TKI는 공유 억제제로 알려져 있다. 이러한 억제 제는 tyrosine kinase의 특정 부위에서 cysteine에 공유 결합하 도록 개발되었다. cysteine 잔기에 존재하는 황 (S)은 전자가 풍부한 원자로, TKI의 친 전자성 그룹과 반응한다. 그 결과, TKI와 cysteine 잔기는 전자를 공유함으로써 비가역적으로 결합한다. 이것은 TKI가 ATP의 tyrosine kinase에 대한 결합 을 차단하고 tyrosine kinase의 활성화를 방지한다 [86,88].
5.3. Tyrosine kinase inhibitor family 5.3.1. Imatinib
다양한 tyrosine kinase inhibitor들이 개발되어 승인을 받았으 며, 그 중 imatinib은 최초의 FDA 승인 TKI로 ABL, BCR- ABL, PDGFRA 및 c-KIT에 대해 활성을 갖는다 (Table 6, Fig. 9). Tyrosine kinase의 활성 부위는 각각 ATP에 대한 결합 부위를 가지고 있고 tyrosine kinase에 의해 촉매 되는 효소 Table 6. Summary of approved tyrosine kinase inhibitor family
Drug Target Target diseases Approval date Company
Imatinib Abl, PDGFR, SCFR CML, GIST 2001 Novartis
Gefitinib EGFR NSCLC 2003 AstraZeneca
Nilotinib Bcr-Abl, PDGFR CML 2004 Novartis
Sorafenib Raf, VEGFR, PDGER Advanced RCC 2005 Bayer
Sunitinib PDGFR, VEGFR, GIST, Advanced RCC 2006 Pfizer
Dasatinib Bcr-Abl, SRC, PDGFR CML 2006 Bristol-Myers Squibb
Lapatinib EGFR Breast cancer 2007 GlaxoSmithKline
Pazopanib VEGFR, PDGFR, FGFR Advanced RCC, STS, NSCLC 2009 GlaxoSmithKline
Crizotinib ALK NSCLC 2011 Pfizer
Ruxolitinib JAK1, JAK2 Myelofibrosis 2011 Novartis
Vandetanib VEGFR, EGFR Advanced Thyroid cancer 2011 AstraZeneca
Axitinib VEGFR Advanced RCC 2012 Pfizer
Bosutinib Abl, SRC CML 2012 Wyeth
Afatinib EGFR NSCLC 2013 Boehringer Ingelheim
Erlotinib EGFR NSCLC 2013 Roche
Ceritinib ALK NSCLC 2014 Novartis
Osimertinib EGFR NSCLC 2015 AstraZeneca
Lenvatinib VEGFR DTC 2015 Eisai
Alectinib ALK NSCLC 2015 Roche
Regorafenib VEGFR, EGFR HCC, CRC, GIST 2017 Bayer
Neratinib HER2 Breast cancer 2017 Puma
Brigatinib ALK NSCLC 2017 Ariad
Tucatinib HER2 Breast cancer 2020 Seattle Genetics
활성은 ATP에서 기질의 tyrosine residues로 말단 인산염을 전달한다 [89]. Imatinib는 ATP 결합 부위에 가깝게 결합하여 폐쇄되는 방식으로 작동한다. 이 과정은 백혈병 발생을 촉진 하는 downstream signal pathway를 “swiching”한다. Imatinib 은 비 암세포의 ABL 단백질도 억제하지만, 일반적으로 세포 에는 ABL tyrosine kinase가 억제되더라도 계속 기능할 수 있 는 추가 중복 tyrosine kinase가 있다. 그러나 일부 종양 세포 는 BCR-ABL에 의존한다. Bcr-Abl에 대한 활성 이외에도 돌 연변이된 c-KIT 및 PDGFR을 억제하기 때문에, 종양의 90%
가 c-KIT 돌연변이를 보유하고 30%~50%가 PDGFR에서 돌 연변이를 보유하는 Gastrointestinal Stromal Tumor (GIST)의 치료에 사용된다 [90]. Imatinib은 GIST에서 발생하는 돌연 변이 c-KIT에 효과적이지만, acute myeloid leukemia (AML) 및 전신 비만세포증 (Systemic mastocytosis, SM)에서 발생하 는 활성 부위 돌연변이에 대해서는 활성이 없다.
5.3.2. Afatinib
Afatinib은 EGFR 및 HER2 표적을 동시에 억제하는 2세대의 대표적인 비가역적 강력한 TKI이다 (Fig. 10). Gefitinib과 같 은 1세대 TKI와는 달리, afatinib은 ErbB 수용체 계열 (EGFR / ErbB1, HER2 / ErbB2, ErbB3, ErbB4)의 모든 관련 동종이 량체 및 이종이량체로부터의 신호를 비가역적으로 차단하 는 ErbB 계열 차단제이다 [91]. ErbB 수용체에 공유 결합하 여 인산화를 차단하고 따라서 downstream 세포 signal 전달 을 차단한다. 이 약물은 2013년 7월에 전이성 NSCLC에서 EGFR 엑손 19 결실 또는 엑손 21 (L858R) 치환 돌연변이에 대한 1차 치료제로 FDA에 의해 승인되었다. Park과 공동연 구자들은 afatinib이 폐의 III기 또는 IV기 선암종 환자에서 무 진행 생존 및 치료 실패 시간을 상당히 개선시켰다고 보 고했다 [92]. 추가 연구에 따르면, Afatinib은 trastuzumab으 로 치료한 후 진행된 HER2- 양성 유방암 환자에서 유망한 활 성을 보였으며 견딜 수 있는 부작용이 있었다 [93]. Afatinib은 gemcitabine과 cisplatin의 조합으로 치료한 그룹과 비교하여
진행된 EGFR 돌연변이 양성 NSCLC의 진행을 상당히 지연 시켰다 [94]. 또한, Afatinib는 EGFR TKI- 민감성 및 내성 세 포주와 NSCLC의 이종 이식 모델 모두에서 더 강력한 항암 활성을 보여주었다 [95].
5.3.3. Sorafenib
Sorafenib은 RAF-1, VGFR-2 및 VGFR-3 및 기타 RTK 활성 을 억제할 수 있다 (Fig. 10) [96]. RAF kinase와 VEGFR kinase 를 동시에 표적으로 하고 억제하는 최초의 항종양제이다 [97]. 직접적으로 RAF / MEK / ERK에 의해 매개되는 세포 신 호 전달 경로를 차단함으로써 암세포의 증식을 억제할 수 있 는 종양 세포의 영양 공급 차단과 또한 VEGFR의 작용을 통한 혈관의 형성을 억제한다. 임상 연구에 따르면 Sorafenib은 신 장암 환자의 무 진행 생존 (PFS)을 상당히 연장시킬 수 있으며, 주요 부작용은 메스꺼움, 설사, 발진 및 고혈압이다 [98].
5.3.4. Pazopanib
Pazopanib은 VEGFR 매개 신생 혈관 형성의 선택적 억제와 성장 촉진 수용체의 직접적인 차단을 통해 항종양 효과를 가 진 경구용 다중 표적 kinase 억제제이다 (Fig. 10) [99]. 혈소 판 유래 성장 인자 수용체 (PDGFR), 섬유아세포 성장 인자 수용체 (FGFR)를 포함한 RTK. 전이성 renal cell carcinoma (mRCC) 치료에 대한 판매 허가를 받았다 [100]. Pazopanib은 진행성 soft tissue sarcoma (STS)의 여러 아형 치료를 위한 허 가된 최초의 kinase 억제제로 이 승인은 pazopanib을 투여받 은 진행성 STS 환자에서 무 진행 생존 (PFS)의 상당한 연장 을 입증한 이중 맹검, 위약 대조 무작위 3상 시험의 결과를 기반으로 한다. 그러나 이러한 항종양 효과에도 불구하고, 전체 생존율 (Overall Survival/OS)에서 유의한 차이가 관찰 되지 않았다.
5.3.5. Osimertinib
Osimertinib은 EGFR-T790M 양성 NSCLC 치료를 위해 2017
Fig. 10. Structure of tyrosine kinase inhibitors [Wikipedia].
년 3월에 승인된 최초의 글로벌 EGFR TKI로 EGFR-T790 돌 연변이를 목표로 하는 새로운 3세대 TKI이다 [101]. EGFR의 T790 돌연변이는 1세대와 2세대 EGFR TKI에 대한 내성 저 항의 대부분을 담당하며 진행된 NSCLC에서 EGFR-양성 돌 연변이의 치료에 중대한 어려움을 나타낸다. 항암 효과 이외 에도, osimertinib은 ABC 수송체의 과발현에 의해 유도된 MDR 을 역전시키는 chemosensitizing agent로서 작용할 수 있다.
5.4. Development of TKI
현재 많은 TKI 약물들은 임상시험이 진행되고 있다. Bruton Tyrosine Kinase (BTK) 억제제인 zanubrutinib은 생화학적 분 석에서 BTK에 대한 전임상 연구에서 유리한 약동학 / 약력 학적 특성을 입증했다 (Table 7) [102]. 임상 1상 시험에서 zanubrutinib은 매일 두 번 160mg으로 치료받은 환자의 혈액 및 림프절 생검에서 완전하고 지속적인 지속적인 24시간 BTK 점유를 보였으며 non-Hodgkin lymphoma 환자에서 지 속적인 반응을 보인다. Poziotinib (HM781-36B)는 EGFR, HER2 및 ErbB4를 표적으로 하는 비가역적 pan-HER TKI이 다 [103]. Poziotinib은 EGFR TKI 내성 폐암 세포를 포함한 다양한 암 세포주가 발현하는 EGFR 또는 HER2에서 우수 한 시험관 내 활성을 보여준다. 2 상 임상 시험에서 Poziotinib 은 ErbB 계열 kinase를 효과적으로 억제했다. 이와 함께 나 노 기술 및 표적 전달을 통해 TKI의 치료 응용을 확대해가 고 있으며, 약물의 효능 개선, 부작용 감소, 생체 이용률 향 상 및 약물 안정성 증가를 통해 다양한 치료 효과를 얻고 있 다 [104].
6. CONCLUSION
많은 화학요법 항암제들이 개발되어 우수한 항암 효과를 보 여왔으며 최근 표적전달 시스템 및 면역항암제들과의 병용 요법으로 지속적인 관심과 개발이 진행되고 있다. 특히 억제 제 약물들은 정상 세포와는 달리 암세포 내에 특이적으로 과 발현되는 단백질을 표적으로 하여 표적 단백질에 결합하고 이의 활성을 억제한다. 이를 통해 과발현된 신호전달에 의한 암세포의 증식 및 전이를 차단하여 항암 효과를 나타내게 된 다. 억제제 약물의 표적 단백질은 정상 세포에서 발현이 낮 기 때문에 기존 화학항암제와 비교하여 정상 세포에 대한 영 향이 낮지만, 약물의 독성이 낮은 단점이 있다. 하지만 최근 억제제 약물의 용해도 개선 및 표적 약물 전달 시스템의 적 용으로 약효를 높이고 있다. 더욱이 최근 antibody-drug conjugate(ADC)에 억제제 약물이 도입되면서 억제제 약물들 의 ADC 개발 관심이 높아지고 있으며, 다양한 억제제의 개 발도 진행되고 있다. 이처럼 다양한 억제제 약물들의 신규 후보물질 개발 및 임상 시험 진행은 새로운 화학항암제가 개 발될 것으로 보이며 암과 함께 많은 질병 치료제로 적용될 것으로 기대된다.
Acknowledgements
This study was supported by National Research Foundation of Korea (NRF, 2017R1C1B5074521, 2019R1F1A1059737).
Figure 1 was designed by Gaeun Choi (Kangwon National Table 7. Tyrosine kinase inhibitor family in clinical trial
Drug Phase NCT# Target diseases Current State Sponsor
ASP8273 I 02113813
A dose escalation study of ASP8273 in subjects with Non-Small- Cell Lung Cancer (NSCLC) who have Epidermal Growth Factor
Receptor (EGFR) mutations
Completed Astellas Pharma Global Development, Inc.
Zanubrutinib I 03465059 Study of the safety and pharmacokinetics of BGB-3111 in subjects
with B-Cell lymphoid malignancies Completed BeiGene
K0706 I 02629692 Safety, tolerability, pharmacokinetics and activity of K0706 Recruiting
Sun Pharma Advanced Research Company
Limited
Poziotinib II 02544997
Single-Arm trial of poziotinib as salvage treatment in patients with metastatic breast cancer who has HER2 or EGFR mutation or
activated AR or EGFR pathway
Completed Samsung Medical Center
Cabozantinib II 03339219Study of cabozantinib in japanese patients with advanced renal cell
carcinoma Completed Takeda
Anlotinib II 02586350 Study of anlotinib in patients with medullary thyroid
carcinoma(ALTER01031) Completed
Chia Tai Tianqing Pharmaceutical Group
Co., Ltd.
Pexidartinib II 01349036 Study of PLX3397 in patients with recurrent glioblastoma Terminated Daiichi Sankyo, Inc.
Asciminib III 03106779Study of efficacy of CML-CP patients treated with ABL001 versus
bosutinib, previously treated with 2 or more TKIs Recruiting Novartis Pharmaceuticals
![Fig. 3. Structure of topoisomerase type 2 inhibitors [8].](https://thumb-ap.123doks.com/thumbv2/123dokinfo/4710287.260976/4.892.77.818.127.492/fig-structure-topoisomerase-type-inhibitors.webp)
![Fig. 4. Overview of HSP90 function in the cancer cells and normal cell (a) and HSP90-interacting co-chaperones and HSP90-dependent client proteins (b) [32,35].](https://thumb-ap.123doks.com/thumbv2/123dokinfo/4710287.260976/5.892.85.817.131.458/overview-function-cancer-normal-interacting-chaperones-dependent-proteins.webp)
![Fig. 5. The HSP90 chaperone cycle (a) and HSP90 biding site with HSP90 inhibitors (b) [32,36]](https://thumb-ap.123doks.com/thumbv2/123dokinfo/4710287.260976/6.892.105.797.813.1079/fig-hsp-chaperone-cycle-hsp-biding-site-inhibitors.webp)
![Fig. 7. mTOR signal pathway in the cell [54].](https://thumb-ap.123doks.com/thumbv2/123dokinfo/4710287.260976/9.892.175.725.688.1082/fig-mtor-signal-pathway-cell.webp)
![Fig. 8. Structure and function of mTOR complex 1 and 2 [64].](https://thumb-ap.123doks.com/thumbv2/123dokinfo/4710287.260976/10.892.124.766.110.331/fig-structure-function-mtor-complex.webp)
![Fig. 9. Summary of signaling mechanism of receptor tyrosine kinase(RTK) [76].](https://thumb-ap.123doks.com/thumbv2/123dokinfo/4710287.260976/12.892.174.726.654.1073/fig-summary-signaling-mechanism-receptor-tyrosine-kinase-rtk.webp)
![Fig. 10. Structure of tyrosine kinase inhibitors [Wikipedia].](https://thumb-ap.123doks.com/thumbv2/123dokinfo/4710287.260976/14.892.113.785.811.1073/fig-structure-of-tyrosine-kinase-inhibitors-wikipedia.webp)

