출생 후 커다란 두혈종과 혈소판감소증을 보인 Jacobsen Syndrome 1예
신주희
전체 글
신주희
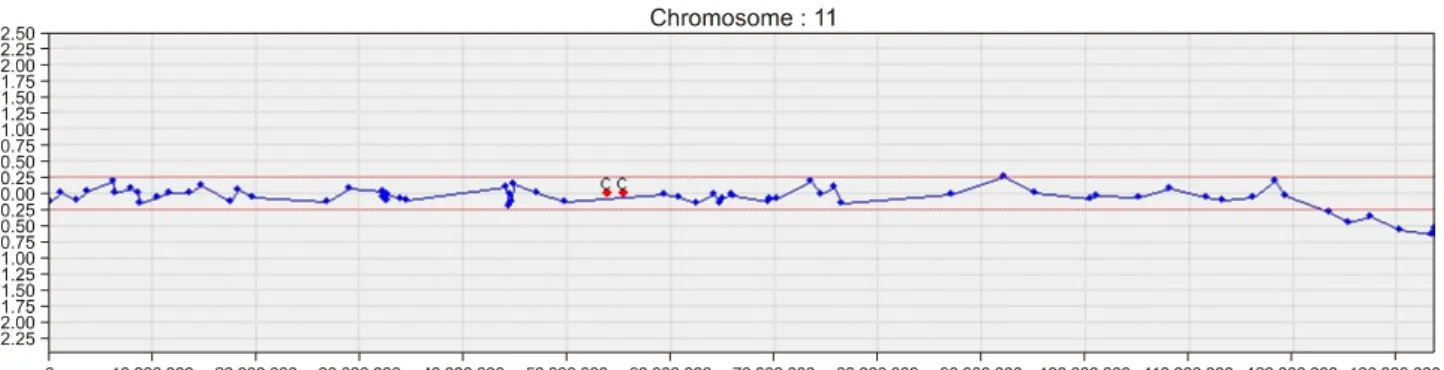
수치

관련 문서
• Normal Giants - These are mainly low-mass stars at the end of their lives that have swelled to become a giant star. This category also includes some high mass stars
All patients with fever, thrombocytopenia and leukocytopenia in SFTS endemic areas should be suspected of SFTS, even they don’t have his- tory of tick bite... Major exposure
Long-term outcome after an early invasive versus selective invasive treatment strategy in patients with non.ST-elevation acute coronary syndrome and elevated
An investigation of adults with Asperger syndrome or high functioning autism, and normal sex differences.. The facilitation of social-emotional understanding
"At the heart of the Iran deal was a giant fiction: that a murderous regime desired only a peaceful nuclear energy program.. Today, we have definitive proof that this
19) Yu Y, Kang J., Clinical studies on treatment of chronic prostatitis with acupuncture
Monosaccharide may be present in the form of Monosaccharide may be present in the form of a linear or ring structure. In solution it is in the form of
(which is linearly independent) similar to above series form with a different r and different coeff... has double root, , Case II –
