Vol. 15, No. 1, May, 2007
□ 증 례 □1)
서 론
멘케스 병(Menkes disease)은 구리 결핍에 의해 발생하는 드문 진행성 신경 퇴행 질환으로, 성염색 체 열성 유전한다. 병과 관련된 유전자는 Xq13에 위치한 ATP7A로 이 유전자의 결함에 의하여 세포 및 장 점막의 구리운반에 관여하는 P type ATPase 의 활성이 저하되며, 환자의 60-70%에서 유전자 이상을 발견할 수 있다. 구리 결핍에 의해 특징적인 모발의 이상, 발달 지연, 경련, 결체 조직의 이상 등
본 논문의 요지는 2007년 제 9차 아세아 오세아니아 소아신 경학회에서 포스터로 발표되었음.
책임저자 : 김은영, 광주기독병원 소아과
Tel : 062)650-5045, Fax : 062)650-5040 E-mail : [email protected]
여러 가지 증상을 나타내며, 대개 예후도 좋지 않아 3세 이전에 사망하므로 추적관찰도 용이하지 않다
1-3). 일부에서 copper-histidine 치료로 구리를 보 충하여 중증도를 감소시키고, 생존 기간을 연장시킨 다는 보고가 있다4). 국내에서 1987년 Moon 등5)이 처음 보고한 이래, 몇 례가(2001년 이후 유전자 진 단 가능6), 2004년 이후 copper-histidine 투여 시도
7)) 보고되었으나, 자연경과를 장기간 추적 관찰한 보고는 없는 실정이다. 이에 저자들은 멘케스 병으 로 진단된 환아를 3년간 추적 관찰하여 그 경과를 보고하는 바이다.
경련과 발달 지연을 주소로 내원한 6개월 남아 로, 갈색의 짧고 꼬이고 거친 모발과 낮은 혈중 구 리, ceruloplasmin 농도를 보여 멘케스 병으로 진단
멘케스 병 환아의 3년 추적관찰
광주기독병원 소아과, 울산대학교 의과대학 서울아산병원 소아과*
유주희·백 현·정 권·선규근·유한욱*·김경심·김용욱·김은영
= Abstr act =
3-year Follow-up of a Menkes Disease Patient
Ju Hee You, M.D., Hyun Paek, M.D., Kwon Jung, M.D.
Gyu Keun Sun, M.D., Han Wook Yoo, M.D.*, Kyoung Sim Kim, M.D.
Yong Wook Kim, M.D. and Eun Young Kim, M.D.
Department of Pediatrics, Kwangju Chistian Hospital, Gwangju, College of Medicine, Ulsan University, Asan Medical Center
*, Seoul, Korea
Menkes disease is a rare fatal X-linked recessive disorder characterized by a gene- ralized defect in intracelluar copper transport. The clinical features which arise from copper deficiency include progressive neurologic deterioration, epilepsy, hair and connective tissue abnormalities. Menkes disease is caused by mutations in the gene encoding the Menkes protein(ATP7A, copper transporting P-type ATPase), which is located on the long arm 13 of the X-chromosome. ATP7A mutations are found in 60 to 70% of the patients. We have experienced a case of Menkes disease in a 6-month-old male who showed develop- mental delay, myoclonic seizures and kinky hair. The serum copper and ceruloplasmin levels were low and the missense mutation(c.3352G>A, resulting in p.G1118S) in exon 17 of ATP7A gene was found. During 3-year follow-up, he regressed developmentally and showed brain atrophy, multiple bladder deverticula, and bony deformities.
Key Words : Menkes disease, Copper, ATP7A mutations
하고, ATP7A 유전자의 돌연변이를 확인하였다. 3 년 추적 관찰 동안 copper-histidine을 단기간 투여 하였으나, 신경학적 퇴행은 진행하면서, 뇌 위축, 다 발성 방광 게실, 골 변형 등이 동반되었기에 문헌고 찰과 함께 보고한다.
증 례
환 아 : 남아, 6개월 주 소 : 경련 및 발달 지연
과거력 : 첫 아이로 재태연령 38주, 체중 3.16 kg 으로 정상 질식 분만하였으며, 신생아 황달로 본원 에 입원한 병력 있었다.
가족력 : 간질이나 발달 지연의 가족력은 없었고, 엄마가 곱슬머리였다.
현병력 : 3주 전부터, 주로 깨어있는 상태에서, 깜 짝 놀라듯 울면서 팔을 앞으로 모으는 양상의 근간 대성 경련을 하루에 1-2회 보였다. 엎어 놓으면 고 개를 잠시 들고, 눈맞춤이 가끔 되나, 옹알이는 하 지 못하는 1개월 수준의 발달을 보였다.
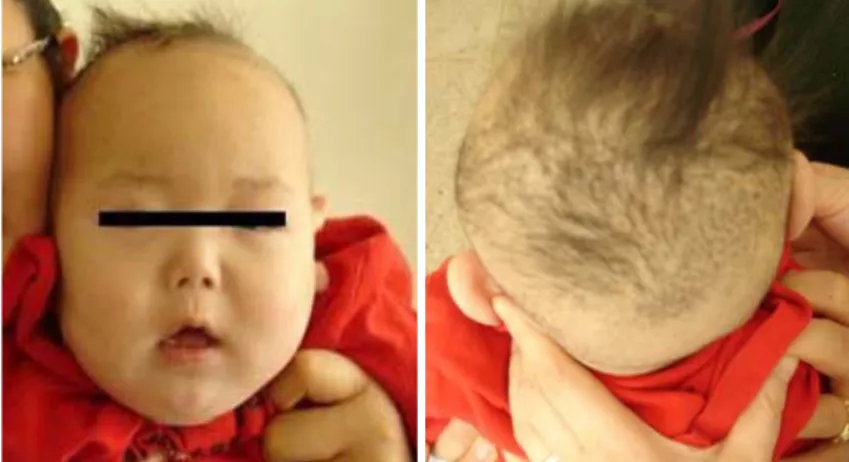
진찰 소견 : 환아의 키는 68 cm(25-50 백분위 수), 체중은 8.5 kg(25-50 백분위수), 두위는 43 cm(25-50 백분위수)이었다. 전반적으로 처져 보였 고, 희고 무표정한 얼굴과 장밋빛 뺨, 연한 갈색의 숱이 적고, 짧고, 꼬이고, 푸석거리는 모발을 보였다
(Fig. 1). 심음과 호흡음은 정상이었고 신경학적 검 사상 전신적인 근력은 III-IV/V 단계로 감소되었 고, 근긴장도는 체간은 저하, 상하지는 증가, 심부건 반사는 항진되었다. 시각, 청각자극에 대한 반응은 정상이었다.
검사 소견 : 말초 혈액 검사상 혈색소 10.2 g/dL, 백혈구 9,300/mm3, 혈소판 282,000/mm3이었다. 일 반 화학검사상 Na+ 139 mEq/L, K+ 4.7 mEq/L, Cl- 103 mEq/L, AST/ALT 25/14 IU/L, 혈중 암 모니아는 48 µmol/L, 혈당은 80 mg/dL이었고 요 검사는 정상이었다. 염색체 검사는 46, XY로 정상 이었고, 혈청 아미노산, 소변 유기산 검사와 혈중 pyruvate, lactate, carnitine치는 정상이었다. 혈청 구리 41.5 ug/dL(정상치 75-145 ug/dL), cerulo- plasmin 12 mg/dL(정상치 16.2-35.6 mg/dL)로 낮 아져 멘케스 병을 의심하였다. 24시간 소변내 구리 배출량도 10.1 ug(정상치 15-30 ug/day)로 적었다.
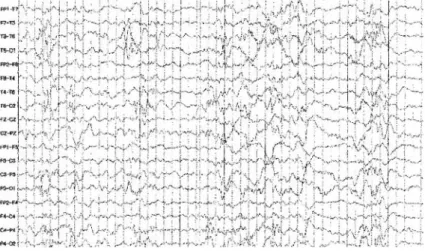
수면 뇌파는 주로 양측 후반부에 다국소성 극파 (multifocal spikes)가 빈번하게 보이면서, 고부정뇌 파(hypsarrhythmia)가 가끔 섞이는 형태로 관찰되 었으며(Fig. 2), 뇌 자기공명영상은 정상이었다.
ATP7A 유전자 검사를 위해, 환아 말초혈액의 백 혈구에서 분리한 DNA를 이용하여 중합효소 연쇄 반응 후 염기서열 분석(PCR sequencing)을 시행하 였다. 17번 exon의 3,352번째 염기 guanine이 ade-
Fig. 1. Facial appearance shows hypopigmented skin, pudgy, rosy cheeks, and brown colored, kinky hair.
nine으로 바뀐 반접합자(hemizygote)로, 1,118번째 아미노산인 glycine이 serine으로 바뀐 missence mutation을 확인하였다. 환아의 엄마는 같은 돌연 변이를 보이는 이형접합자(heterozygote)로 보인자 상태였다(Fig. 3).
치료 및 경과 : 근간대성 경련은 topiramate(9 mg/kg/day) 투여로 10개월 이후부터 소실되었으 나, 재활치료 후에도 전신적인 근력, 근긴장도의 저 하를 포함한 신경학적 증상은 호전되지 않았다. 10 개월에 혈청 구리는 14.3 ug/dL, ceruloplasmin 12 Fig. 3. DNA PCR sequencing using peripheral leukocyte: ATP7A
mutation shows G to A transversion in codon 1118, causing substitution of serine for glycine in exon 17.
Fig. 2. Sedated sleep EEG shows frequent multifocal spikes and oc- casionally, short spike wave bursts with depressed background activity are also shown.
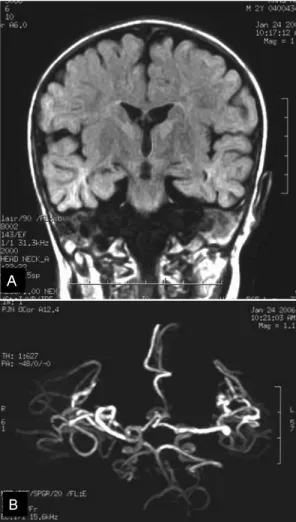
mg/dL로 진단시보다 더 감소되었다. 15개월부터 copper-histidine(757 ug/uL) 100uL를 매일 경피 주사하여, 모발의 색도 진해지고 꼬임과 거침도 호 전되었으나, 한 달 후 환아가 식사량이 줄고 보채며 힘들어 하여 엄마가 임의로 투약을 중단하였다. 그 후로도 반복적인 호흡기 감염과 중이염, 수두, 수족 구병 등으로 자주 입원하였다. 23개월에 추적한 뇌 자기공명영상에서 대뇌와 소뇌는 전반적으로 위축 되어 있었고(Fig. 4A), 뇌 자기공명 혈관 조영술검 사에서 꼬여있고 연장된 뇌동맥들을 확인하였다 (Fig. 4B). 잦은 요로감염으로 인해 시행한 방광조
영술에서 다발성 방광 게실을 같은 시기에 진단하 여(Fig. 5), 감압 목적으로 방광조루술(cystostomy) 을 시행하였다. 3세경, 체중 9 kg(<3 백분위수), 키 80 cm(<3 백분위수), 목가누기는 여전히 안되고, 가끔 눈맞춤만 가능한 정도의 심한 성장발달지연과 골 변형으로 생각되는 요골 하부 말단의 돌출을 보 였다.
고 찰
멘케스 병은 1962년 Menkes 등8)이 한 가족 내 다섯 명의 남아가 신경학적 퇴행, 독특한 머리카락, 심한 성장지연(failure to thrive)을 보이는 경우를 보고하면서 처음 알려졌다. 발생 빈도는 1/100,000- 1/300,000 정도로 매우 드물고, 대개 성염색체 열성 으로 유전하나, 1/3은 가족력 없이 새로운 돌연변이 에 의해 발생한다. 멘케스 병의 원인 유전자는 Xq 13.3에 위치, 23개의 exon과 150 kbp의 길이를 가 지며, 1,500개의 아미노산으로 이루어진 copper- transporting P-type ATPase(ATP7A)를 암호화 하고 있다2). ATP7A 유전자 이상은 환자의 60-
Fig. 4. (A) Brain MRI(T1-weighted image, coronal view) at the age of 23 months shows diffuse cere- bral atrophy. (B) Axial image of brain MR angio- graphy presents tortuous and elongated intracranial
vessels. Fig. 5. Cystography shows a huge diverticulum
on the right side of bladder.
A
B
70%에서 발견되는데, 이상의 70-80%는 점 돌연변 이, 20%는 southern blot으로 진단되는 정도의 비 교적 큰 결실, 1%는 염색체 검사로 발견되는 이상 의 형태를 보인다. 돌연변이의 형태는 다양하여 small insertions/deletions(39%), splicing defects (23%), nonsence mutations(20%), missence mu- tations(17%) 등으로 중합효소 연쇄반응으로 진단 할 수 있다1). 본 증례의 경우 ATP7A 유전자 검사 를 위해 환아의 말초혈액 백혈구에서 분리한 DNA 를 이용하여 PCR sequencing을 시행하였다. Exon 17의 1,118번째 아미노산인 glycine이 serine으로 바뀐 missence mutation을 보였으며, 이는 새로운 변이이나, 유사한 형태(p.G1118D)가 보고된 바 있 고, 이 부위는 ATP7A 단백의 변형을 야기한다고 알려졌다9). 환아의 어머니는 같은 돌연변이를 보이 는 보인자로 부석거리는 곱슬머리 외에는 멘케스 병의 임상증상은 없었다. 유전자 검사를 통하여 성 염색체 열성 유전의 가계임을 확인하였다.
Copper transporting P type ATPase(ATP7A) 는 간을 제외한 모든 조직의 세포막이나 골지체에 위치하여 구리의 세포내 이동 및 세포외로의 유출 에 관여하는데, 결함이 있는 멘케스 병의 경우, 장 점막의 구리 흡수 장애가 생기고 일부 흡수된 구리 도 세포 내에서 골지체로 전달이 안되어 구리 의존 성 효소들의 결함이 발생되며, 혈액으로의 유출 장 애도 생긴다10). 따라서 장, 신장, 비장, 근육, 배양된 섬유아세포에는 구리가 축적되나, 간, 혈액, 뇌에서 는 구리의 결핍이 일어나고, ceruloplasmin 합성과 구리 의존성 효소들의 활성이 저하된다. 특히 구리 의존성 효소들의 활성 저하가 멘케스 병의 다양한 임상증상들과 관련되어 있는데, Dopamine β- hydroxylase(catecholamine 합성), cytochrome c oxidase(사립체내 ATP 합성), superoxide dismu- tase(free radical detoxification), peptidylgycine monooxygenase(removal of the carboxy-terminal glycine residue from neuroendocrine precursors) 의 결함은 심한 신경계 변성을 초래한다. Lysyl oxidase의 결핍은 결체 조직의 합성 장애를 일으켜 약하고 늘어지며 꼬인 혈관, 방광 게실, 위장관 용
종, 탈장을 발생시키는데 다른 효소들보다 결핍이 더 심하다. Sulfhydryl oxidase, monoamine oxidase 의 결핍은 염전모(pili torti)를 tyrosinase, catechol oxidase의 결핍은 피부와 모발의 저색소증을 초래 한다. Ascorbate oxidase는 골격계 결함과 관련이 있다1, 2).
멘케스 병 환아는 대개 정상으로 태어나고(1/3은 미숙아 또는 저체중아로 출생), 신생아기에 수유 곤 란, 저체온, 호흡곤란, 저혈당, 황달, 두혈종 등의 비 특이적인 증상을 보이므로 이시기에 진단하기는 무 척 어렵다3, 11). 신경변성에 의해 나타나는 발달지연 및 퇴행, 경련 등의 신경학적 이상은 생후 3개월 이 후에야 나타나는데, 진단시 고개 가누기가 안되고, 체간의 근긴장도는 심하게 저하되며 심부건 반사는 항진되는 모습을 보인다3). ‘Kinky hair disease’,
‘steely hair disease’로 불릴 정도로 모발의 이상은 모든 환자에서 나타나 진단에 중요한 단서가 된다.
출생시 정상이었다가, 시간이 지나면서 색깔은 연해 지고 꼬이고 거칠고 짧으면서 쉽게 끊어지는 머리 카락으로 변하고, 숱이 적어지며, 머리의 측면과 뒤 쪽에서 더 심하다. 현미경으로 관찰하면 모발이 180°꼬여있는 염전모(pili torti)과 횡축으로 결절 이 생기는 열모증(trichoclasis), 장축으로 갈라지는 털분리증(trichoptilosis) 등이 보인다6). 15% 정도 에서는 갈색이나 회색의 꼬인 머리카락이 출생 당 시 또는 1개월 이내 관찰되기도 하므로, 세심한 관 찰로 조기진단을 증가시킬 수 있겠다11). 얼굴은 무 표정하고 창백하며, 뺨과 귀가 늘어져 보이고, 높은 구개를 보이고, 치아의 발생이 지연된다. 시간이 지 날수록 발달은 퇴행하고 심한 성장부진이 동반되는 데, 체중과 머리둘레는 작지만, 키는 유지되는 비대 칭 형태를 취한다1, 2, 5). 간질은 대개 생후 3개월 이 후에 근간대성 경련의 형태로 주로 나타난다고 알 려진 정도였는데, 최근 12명의 환아를 대상으로 한 연구에서 초기(평균 3개월)에는 열과 동반된 국소 성 간대성 간질 중첩증, 그 후에는 영아 연축, 2세 이후에는 다국소 발작, 강직성 연축, 근간대성 경련 등의 다양한 형태를 보이며, 대부분 난치성임을 보 고하였다. 뇌파에서는 다국소성 극서파, 고부정뇌파
(hypsarrhythmia) 등을 보인다12). 또한 lysyl oxi- dase 결핍에 의해 결체 조직의 이상들이 발생하는 데 늘어진 피부, 제대 및 서혜부 탈장, 방광 게실, 뇌 혈관의 꼬임 및 동맥류 등 다양한 형태를 보인
다3, 4, 13). 특히 방광게실은 가장 흔히 발생되는 요로
계 이상으로 1세 이후에 발생하고 연령이 증가할수 록 게실의 빈도 및 이차적인 합병증(요로감염, 방광 결석)의 발생도 증가한다14). Copper-histidine 치료 를 하더라도 요로계 이상의 발생은 막지 못하며, 드 물게 방광 게실 파열에 의한 복막염으로 환자가 사 망하기도 하므로 요로계 이상에 대한 조기 진단 및 그에 따른 적절한 처치가 중요하다 하겠다13). 골격 계의 변화로 골다공증과 골간단의 이형성(metaphy- seal dysplasia)에 의한 주로 장골, 갈비뼈의 말단에 골절 및 돌기(spur)가 생기고 두개골의 보름골 (wormian bone) 형성도 관찰된다. 안과적으로는 시선의 고정과 추적의 이상, 근시, 사시가 흔히 생 기고, 망막의 저색소성과 혈관의 꼬임이나 시신경 위축, 백내장 등도 동반된다. 예후는 좋지 않아 대 부분 3세 이전에 호흡기 부전으로 사망한다1, 2). 저 자들의 증례도 특징적인 모발소견으로 멘케스 병을 의심하였고, 6개월에 발생한 근간대성 경련은 다행 히 항경련제 투여에 의해 잘 조절되었다. 생후 23개 월에 진단한 다발성 방광게실은 감압을 위해 방광 조루술을 시행하였고, 잦은 요로감염이 동반되어 간 헐적 항생제 치료를 병행하고 있다. 3세경 육안적으 로 요골 말단부 돌출이 보여 골변형도 동반되었을 것으로 추측된다. 위에서 언급한 전형적이고 심한 경우 외에도 경한 형태로 생각되는 ‘occipital horn 증후군’이 보고되는데 외견상 후두골에 근육 닿는 부위의 석회화로 인한 돌출(occipital horn)이 특징 적으로 현저한 lysyl oxidase 결핍에 의한 결체조 직 이상이 주가 되며, 신경학적 퇴행은 경하다. 이 경우 ATP7A의 splicing defect가 주로 발견되고, 남아있는 ATPase의 활성도는 2-5% 정도이다4, 10,
13, 15)
.
멘케스 병은 위에서 기술한 특징적인 임상 증상 을 보이면서 혈청 구리 및 ceruloplasmin 농도가 낮은 경우 진단할 수 있다. 대개 진단당시 혈청 구
리는 30 ug/dL(정상치 70-102), ceruloplasmin은 15 mg/dL(정상치 18-35) 이하로 감소되어 있다3). 확진은 피부 섬유아세포를 배양하여 세포 내 구리 의 축적을 증명해야 하는데, 환자의 60-70%는 ATP7A 유전자의 돌연변이가 동반되므로 말초혈을 이용하여 이를 확인하는 것도 확진에 도움이 된다.
출생 후 1개월까지는 정상적으로 혈중 구리(9-46 ug/dL)와 ceruloplasmin(5-26 mg/dL)이 낮은 농 도를 보이므로 신생아에서 의심되는 경우 ATP7A 유전자 분석이나 피부 섬유아세포 배양에 의해 구 리농도를 측정하는 것이 진단에 유용하다. 또한 혈 중 catecholamine의 측정이 유용한데, 이는 구리 의존성 효소인 dopamine β-hydroxylase의 기능 이상이 태아기나 신생아기의 혈중 catecholamine의 농도에 이상을 일으키기 때문이다1-3). 뇌 자기공명 영상에서는 전반적인 뇌 위축, 백질 이상이 보이며, 자기공명 혈관 조영술(MRA)상 꼬불꼬불하게 연장 되어 있는 내경 동맥, 대뇌 동맥과 그 분지가 특징 적이다. 이러한 특징적인 MRA 소견은 전형적인 증상이 나타나지 않는 신생아기에도 관찰되어 조기 진단의 실마리가 될 수 있다16). 병리학적으로 대뇌 피질, 해마, 기저핵, 시상, 소뇌의 과립층, 가시층 (purkinje cell layer)의 신경세포 소실과 성상교세 포증, 백질의 탈수초화 등이 관찰되며5, 13), 뇌혈관들 은 결체조직이 얇고 탄력막(elastic lamina)이 파열 되어1), 경막하 혈종, 출혈과 경색 등이 잘 동반된다
16). 멘케스 병에서 보이는 연한 색깔의 꼬인 모발은 argininosuccinic aciduria2), 모발유황이영양증(tri- chothiodystrophy)17), 외배엽 이형성 증후군18) 등에 서도 발견되므로 감별이 필요하다. 저자들의 증례와 같이 ATP7A 유전자 검사를 시행하여 돌연변이가 있고, 엄마가 보인자이고 성염색체 열성으로 유전하 는 가계로 밝혀진 경우 유전상담이 중요하다. 다음 아이를 임신하여 남아라면 50%에서 멘케스 병 환 자가 되므로 ATP7A 유전자 검사를 시행해야 한 다. 유전자 이상이 밝혀지지 않은 경우는 임신 1기 에 융모막 융모에서 임신 2기에는 양수세포를 배양 하여 축적된 구리농도를 측정하여 산전 진단한다.
보인자인 경우, 임상증상은 없고 일부에서 꼬인 머
리카락과 저색소성 피부를 나타내며, 배양된 피부 섬유아세포에서 구리축적을 보인다고 하나, random X inactivation에 의해 구리축적이 증명되 지 않는 경우도 있으므로 무엇보다 유전자 검사를 시행하는 것이 중요하겠다1, 19).
멘케스 병의 치료는 항경련제 투여, 재활 치료 등 보존적 치료 외에는 확실한 치료법이 없는 실정 이나, copper-histidine을 경피로 투여(elemental copper 50-150 ug/kg/day)하여 구리를 보충하는 방법이 시도되면서 비교적 효과적인 것으로 알려져
있다4, 13). Copper-histidine 투여로 간의 구리 결핍
이 교정되고, 혈청 내 구리와 ceruloplasmin 농도가 정상화되며, 모발의 저색소성과 꼬임도 호전된다.
우리 환아도 copper-histidine 한달 투여 후 모발의 호전을 관찰할 수 있었다. 그러나 신경학적 퇴행에 대한 효과는 확실치 않다. 증상 발현 전 조기 cop- per-histidine 투여로 신경학적 퇴행을 예방하고 생 존 기간을 연장할 수 있다는 보고가 있는 반면4, 13), 효과가 없었다는 보고도 있어20), 개인의 잔존 ATPase 활성도가 예후에 가장 중요한 인자로 생 각된다. 치료시기도 중요한데, 퇴행성 신경변화가 일어나기 전, 산전19) 혹은 출생 2개월 이내에 cop- per-histidine을 투여하면, ATPase의 활성도가 일 부 보존되어 있는 환아의 경우 신경변성이 예방되 거나, 발현이 지연되면서 경한 양상을 나타낸다4, 13,
21). 신경학적 증상이 발현된 이후에 치료를 시작한 경우는 경련 및 보챔과 수면장애를 완화시키고, 근 긴장도 및 운동기능을 호전시키는데 도움이 된다고 는 하나, 궁극적으로 진행하는 퇴행을 막지는 못한
다7, 22). 또한 지속적인 사용으로 인한 구리의 축적
은 적혈구에서 용혈에 의한 혈색소의 누출, 콩팥의 세뇨관 손상 등 다양한 조직에서의 독성을 유발한 다22). 따라서 이미 퇴행성 신경변성이 발생한 환아 에서 copper-histidine 치료를 할 것인지는 논란의 여지가 있다. 결체조직 이상은 조기 치료한 경우라 도 예방하거나 호전시키지 못하며, 이는 copper- histidine 투여가 lysyl oxidase의 활성을 호전시키 지 못하기 때문이다4, 10, 13). 앞으로 멘케스 병의 예 후를 향상시키기 위해, 조기 진단과 뇌 및 구리 의
존성 효소에 구리를 효과적으로 전달하기 위한 방 법의 개발이 필요하다 하겠다.
요 약
저자들은 특징적 모발 소견과 발달 지연, 경련을 보인 6개월 남아에서 낮은 혈청 구리와 ceruloplas- min 농도로 멘케스 병을 진단하고, ATP7A 유전자 의 exon 17에서 missence mutation(p.G1118S)을 확인하였다. 3년 추적관찰 동안 발달은 퇴행하면서 뇌 위축, 다발성 방광게실, 골변형 등이 동반된 1례 를 경험하였기에 문헌 고찰과 함께 보고하는 바이 다.
References
1) Chen H. Atlas of genetic diagnosis and coun- seling. Totowa: Humana Press 2006:639-43.
2) Boustany RN, Zucker A. Menkes' disease. In : Swaiman KF, Ashwal S, Ferriero DM. Pediatric neurology, principles & practice. 4th ed. Phila- delphia: Mosby Elsevier co 2006:1321-6.
3) Komada H, Murata Y, Kobayashi M. Clinical manifestations and treatment of Menkes dis- ease and its variants. Pediatr Int 1999;41:
423-9.
4) Christodoulou J, Dankes DM, Sarkar B, Baer- locher KE, Casey R, Horn N, et al. Early treatment of Menkes disease with parenteral copper-histidine: long-term follow-up of four treated patients. Am J Med Genet 1998;76:
154-64.
5) Moon HR, Chi JG, Yeon KM, Suh YL, Sung RH, Kim BI, et al. Menkes disease-autopsy case with metal analysis of hair. J Korean Med Sci 1987;2:75-84.
6) Lee YS, Park SW, Cha BH, Lim BK, Kim JS, Lee WS, et al. A case of kinky hair disease.
J Korean Child Neurol Soc 2001;9:164-71.
7) Choi JH, Yoo HW. Long term treatment of copper-histidine in a Menkes disease patient.
J Korean Inherit Metab Dis 2004;4:34-9.
8) Menkes JH, Alter M, Steigleder GK, Weakley DR, Sung JH. A sex linked recessive disorder with retardation of growth, perculiar hair, and
focal cerebral and cerebellar degeneration. Pe- diatrics 1962;29:764-79.
9) Hahn S. Identification of four novel mutations in classical Menkes disease and successful prenatal DNA diagnosis. Mol Genet Metab 2001;73:86-90
10) Voskoboinik I, Camakaris J. Menkes copper- transporting P-type ATPase: biochemical and cell biology properties, and role in Menkes disease. J Bioenerg Biomembr 2002;34:363-71.
11) Gu YH, Kodama H, Shiga K, Nakata S, Yana- gawa Y, Ozawa H. A survey of Japanese pa- tients with Menkes disease from 1990 to 2003:
incidence and early signs before typical symp- tomatic onset, pointing the way to earlier diagnosis. J Inherit Metab Dis 2005;28:473-8.
12) Buisson NB, Kaminska A, Nabbout R, Barnerias C, Desguerre I, Lonlay PD, et al. Epilepsy in Menkes disease: analysis of clinical stages.
Epilepsia 2006;47:380-6.
13) George DH, Casey RE. Menkes disease after copper histidine replacement therapy: case report. Pediatr Dev Pathol 2001;4:281-8.
14) Zaffanello M, Maffeis C, Fanos V, Franchini M, Zamboni G. Urological complications and copper replacement therapy in childhood Menkes syndrome. Acta Pediatr 2006;95:785-90.
15) Møller LB, Tümer Z, Lund C, Petersen C, Cole T, Hanusch R, et al. Similar splice-site muta- tions of the ATP7A gene lead to different phe-
notypes: classical menkes disease or occipital horn syndrome. Am J Hum Genet 2000;66:
1211-20.
16) Geller TJ, Pan Y, Martin DS. Early neuro- radiologic evidence of degeneration in Menkes’
disease. Pediatr Neurol 1997;17:255-8.
17) Liang C, Morris A, Schlucker S, Imoto K, Price VH, Menefee E. Structural and molecular hair abnormalities in trichothiodystrophy. J Invest Dermatol 2006;126:2210-6.
18) Jones KL. Smith's recognizable patterns of human malformation. 6th ed. Philadelphia: El- sevier, 2006:628-45.
19) Gu YH, Kodama H, Sato E, Mochizuki D, Yanagawa Y, Tagayanagi M, et al. Prenatal diagnosis of menkes disease by genetic ana- lysis and copper measurement. Brain Dev 2002;24:715-8.
20) Kaler SG, Buist NR, Holmes CS, Goldstein DS, Miller RC, Gahl WA. Early copper therapy in classic menkes disease patients with a novel splicing mutation. Ann Neuol 1995;38:921-8.
21) Kim BE, Smith K, Petris MJ. A copper treatable Menkes disease mutation associated with de- fective trafficking of a functional Menkes cop- per ATPase. J Med Genet 2003;40:290-5.
22) Sheela SR, Latha M, Liu P, Lem K, Kaler SG.
Copper replacement treatment for symptomatic Menkes disease: ethical considerations. Clin Genet 2005;68:278-83.