상호정보와 부울대수를 이용한 복합질환의 SNP 상호작용 예측
1)임 상 섭*, 위 규 범**
Prediction of SNP interactions in complex diseases with mutual information and boolean algebra
Sangseob Leem*, Kyubum Wee**
요 약
대부분의 만성질환은 다수의 유전자-유전자 사이의 상호작용에 의해서 발병하는 복합질환이다. 복합질환의 발 병에 관여하는 단일염기다형성(single nucleotide polymorphism; SNP)과 유전자 사이의 상호작용을 찾아내는 연구는 질환의 예방과 치료에 기여한다. 기존의 연구 방법은 주로 특정 유전자 내 SNP 조합을 찾아내는 데 그치고 있다. 본 연구에서는 SNP 조합의 구성원 사이에 일어나는 구체적인 상호작용을 나타내는 부울식을 찾는 방법을 제 시한다. 본 논문에서 제안하는 방법은 두 단계로 이루어진다. 제 1 단계에서는 엔트로피에 기반한 상호정보를 이용 하여 발병에 관여하는 SNP 조합을 찾는다. 제 2단계에서는 제 1 단계에서 찾은 SNP 조합으로 이루어지는 부울식 중에서 발병 예측정확도가 가장 높은 부울식을 찾는다. 제안한 방법을 임상자료에 적용하여 그 효율성을 실험하였 으며 기존 연구들과 장단점을 비교하였다.
Abstract
Most chronic diseases are complex diseases which are caused by interactions of several genes.
Studies on finding SNPs and gene-gene interactions involved in the development of complex diseases can contribute to prevention and treatment of the diseases. Previous studies mostly concentrate on finding only the set of SNPs involved. In this study we suggest a way to see how these SNPs interact using boolean expressions. The proposed method consists of two stages. In the first stage we find the set of SNPs involved in the development of diseases using mutual information based on entropy. In the second stage we find the highest accuracy boolean expression that consists of the SNP set obtained in the first stage. We experimented with clinical data to demonstrate the effectiveness of the proposed method. We also compared the differences between our method and the previous results on the SNP associations studies.
∙제1저자 : 임상섭 교신저자 : 위규범
∙투고일 : 2010. 09. 01, 심사일 : 2010. 09. 11, 게재확정일 : 2010. 10. 11.
* 아주대학교 박사과정 ** 아주대학교 정보컴퓨터공학부 교수
※ 이 논문은 2006년도 정부(과학기술부)의 재원으로 한국과학재단의 지원을 받아 수행된 연구임.
(No. R01-2006-000-10775-0)
▸Keyword : 복합질환(complex diseases), 단일염기다형성(single nucleotide polymorphism;
SNP), 부울식(boolean expressions), 엔트로피(entropy), 상호정보(mutual information)
Ⅰ. 서 론
복합질환(complex disease)은 여러 유전자들의 복합적인 상호작용(interaction)에 의하여 발병하는 질환을 뜻한다.
많은 사람들이 고통 받고 있는 고혈압, 당뇨, 천식, 비만 등 대부분의 만성질환은 복합질환으로서, 특정한 복합질환의 발 병에 관여하는 유전자들을 밝혀내는 것은 매우 중요하고 의미 있는 작업이다. 발병에 관여하는 유전자들을 밝혀냄으로서 그 질환의 예방 및 치료에 기여할 수 있다.
유전자-유전자 상호작용 및 유전자-환경요인 상호작용이 라는 복잡성으로 인하여 발병에 관여하는 유전자들을 찾아내 는 작업은 많은 어려움이 따른다. 주로 쓰이는 방법은 유전정 보의 개인차를 나타내는 단일염기다형성(single nucleotide polymorphism; SNP)과 질병의 연관성을 분석하는 SNP 연관성 연구(SNP association study)이다.
SNP 연관성 연구에는 여러 가지 통계학적인 방법들과 기 계학습(machine learning)의 기법들이 사용되고 있다[1].
대표적인 기법들을 살펴보면 다음과 같다. 베이지안 추론을 사용하는 BEAM[2], 모든 가능한 SNP의 상호작용을 전체 탐색하는 MDR[3], 로지스틱 회귀분석(logistic regression) 을 이용하는 방법[4], 조합론적 분할 방법을 사용하는 CPM(
combinatorial partitioning method)과 RPM(restricted partition method)[5, 6], 로직 회귀분석과 MCMC(Markov Chain Monte Carlo)를 혼합한 방법[7], 하플로타입 (haplotype) 정보와 랜덤 포레스트(random forest)를 사 용하는 HapForest[8], 결정트리에 기반한 MegaSNPHunter[9]
등의 다양한 방법이 연구되어 있다.
위에서 열거한 방법의 대부분은 복합질환의 발병에 관여하 는 SNP들을 찾아냄으로서 그 SNP들 사이에 상호작용이 존 재함을 알 수 있으나 구체적으로 어떠한 상호작용인지를 설명 하지 못한다. 상호작용을 보여주는 경우에도 그 식이 너무 길 고 복잡하여 이해하기 어렵다.
본 연구에서는 부울식(boolean expression)을 사용하여 상호작용을 표현하고 발병에 관여하는 SNP들을 상호정보 (mutual information)을 이용하여 규명하는 복합적인 접근 법을 제시한다.
본 논문의 구성은 다음과 같다. 제 2 장에서는 상호정보량 과 부울식에 대해서 소개하고 본 연구에서 제안하는 알고리즘
을 설명한다. 제 3 장에서는 임상자료에 적용한 결과를 분석 한다. 제 4 장에서는 기존의 연구 방법들과 비교하고 제안한 방법의 효율성을 분석한다. 제 5 장은 결론과 향후 연구주제 를 제시한다.
II. 방법
본 연구에서 제안하는 방법은 두 단계로 이루어진다. 첫 단계에서는 상호정보를 이용하여 발병에 관여하는 SNP의 집 합을 찾아내고 다음 단계에서 SNP들의 상호작용을 나타내는 부울식을 찾는다.
1. 상호정보 (mutual information)
랜덤변수(random variable) 의 엔트로피(entropy)
는 다음과 같이 정의 된다.
∈
log
엔트로피는 랜덤변수에 담겨 있는 정보의 양을 나타낸다.
두 랜덤변수 , 의 결합 엔트로피(joint entropy)
는 다음과 같이 정의 된다.
∈
∈
log
랜덤변수가 주어졌을 때 랜덤변수의 조건부 엔트로 피(conditional entropy)는 다음과 같이 정의 된다.
∈
∈
log
, , 사이에는 등식
가 성립한다. 두 랜덤변수 의 상호정보는 다음과 같이 정의 된다.
상호정보는와가 공유하는 정보의 양을 나타낸다.
본 연구에서는 각 SNP의 값에 따라 데이터를 이루는 사람 들의 집합가 파티션 되므로, 엔트로피를 다음과 같이 파티 션을 사용하여 정의한다.
⋯ 이 집합의 파티션(partition)이 라고 하자. 다시 말해서,∪∪ ⋯이고 서로 다 른,에 대해서∩ ∅이다. 이때 파티션의 엔트 로피(entropy)는 다음과 같이 정의 된다[10].
log
결합 엔트로피, 조건부 엔트로피, 상호정보도 같은 방식으 로 파티션을 사용하여 정의한다. 여기서 등의 기호 는 집합의 원소의 개수를 뜻한다.
예를 들어서가 환자군(case)과 대조군(control)의 두 부 분으로 이루어지는 파티션이라 하고, 는 특정 SNP인 ALOX5_p1_1708_G>A의 값(GG, GA, AA)에 의해서 세 부분 으로 이루어지는 파티션이라 하자. 이때 는 ALOX5_p1_1708_G>A와 환자군/대조군 분류가 공유하는 정 보량을 나타낸다.
상호정보는 다수의 파티션에 대해서 확장하여 정의할 수 있다[10].
⋯
⋯
⋯
SNP1, SNP2, …, SNPk으로 이루어지는 파티션을 각각
⋯라 하고를 환자군/대조군의 파티션이라 할 때⋯는 SNP1, SNP2, …, SNPk의 상 호작용과 질환 발병의 연관성을 나타낸다.
상호정보의 값은와의 연관성에 의해서 도 영향을 받지만 와 의 자체 엔트로피 값 와
에 의해서도 영향을 받는다. 따라서 본 연구에서는 와 의 연관성을 나타내기 위해 를
로 나눈 정규상호정보 (normalized mutual information) 을 사용한다[10].
⋯
⋯
⋯
2. 부울 식(boolean expression)
서론에서 언급한 바와 같이 대부분의 기존 SNP 연관성 연 구는 발병에 관여하는 SNP의 조합을 찾아내지만 어떠한 상
호작용인지 설명하지 못한다. 본 연구에서는 상호작용을 부울 식으로 표현하여 상호작용의 생물학적 의미를 유추할 수 있는 단서를 제공한다.
예를 들어 논리곱(conjunction)으로 표현되는 식은 두 개 의 SNP이 동일한 경로(pathway)에 존재하거나 동일한 유 전자(gene)상에 존재하는 경우를 나타낼 수 있고, 논리합 (disjunction)으로 표현되는 식은 두 SNP이 독립적으로 발 병에 관련이 있는 작용을 나타낼 수 있다. 부정(negation)으 로 표현되는 식은 질병에 저항하는 작용 등을 나타낼 수 있다.
SNP의 유전형(genotype)은 우성/우성 대립유전자 (major/major allele), 우성/열성 대립유전자(major/minor allele), 열성/열성 대립유전자(minor/minor allele)의 세 가지이다. SNP의 유전형에 따라 SNP의 표현형(phenotype) 이 어떻게 결정되는가는 우성모델(dominant model), 열성 모델(recessive model), 혼합형모델(codominant model) 이 있다. 우성모델(dominant model)은 우성 대립유전자의 유무에 의해서 표현형이 결정되는 것이며, 열성모델(recessive model)은 열성 대립유전자의 유무에 의해서 표현형이 결정 된다. 혼합형모델(codominant model)은 세 가지의 유전형 이 각각 세 가지의 다른 표현형(phenotype)으로 나타나는 모델이다.
예를 들어서, 어떤 SNP이 G가 우성이고 A가 열성이라고 하자. 이 SNP의 유전형은 GG, GA, AA의 세 가지가 있다.
이때 이 SNP이 우성모델이라는 것은 다음을 의미한다. 유전 형이 GG 와 GA 인 경우의 표현형이 같으며, 유전형이 AA인 경우의 표현형과는 다르게 나타난다. 유전형이 GG 또는 GA 이면 갈색머리이며 유전형이 AA 이면 금색머리인 경우를 생 각할 수 있다. 다시 말해서 우성 대립유전자인 G 의 유무에 의해서 표현형이 결정되는 것을 의미한다.
이 SNP이 열성모델이라는 것은 다음을 뜻한다. 유전형이 GA와 AA인 경우의 표현형이 같으며, 유전형이 GG인 경우 의 표현형과는 다르게 나타난다. 유전형이 GA 또는 AA 이면 갈색머리이며 유전형이 GG이면 금색머리인 경우를 생각할 수 있다. 다시 말해서 열성 대립유전자인 A의 유무에 의해서 표현형이 결정됨을 뜻한다.
본 연구에서는 각 SNP이 우성모델 또는 열성모델로 나타난다 고 가정했으며 SNP의 상호작용을 나타내는 부울식을 탐색하는 것과 더불어 각 SNP이 우성모델인지 열성모델인지도 탐색한다.
3. 알고리즘
본 연구에서 제안하는 방법은 제 1 단계로 정규상호정보가 높은 SNP 조합을 찾아내고 제 2 단계로 각 조합에 속한
SNP 들로 이루어진 부울식 중에서 발병 예측정확도가 높은 식을 찾는다. 제 1 단계에서는 k 개로 이루어지는 SNP의 모 든 조합에 대해서 normalized mutual information을 계산 하여 이중 상위 20위 까지의 SNP 조합을 선정한다. 제 2 단 계에서는 각 SNP 조합으로 이루어지는 모든 부울식을 생성 하고 각 부울식의 발병 예측정확도를 측정한다. 이 중에서 예 측정확도가 가장 높은 부울식을 선정한다.
기존의 SNP 상호작용 연구들은 대부분 5개 이하 조합을 대상으로 하므로, 본 연구에서도 SNP의 2개 조합, 3개 조합, 4개 조합, 5개 조합의 경우만 조사하였다. 그러나 본 연구에 서 제안하는 방법은 6개 이상의 SNP 조합의 경우로도 확장 가능하다.
그림 1. 제안한 방법의 진행 순서 Fig. 1. Procedure of proposed method
각 SNP 조합과 발병의 연관성을 나타내는 정규상호정보 (NMI)는 제 2.1 절에서 기술한 식을 사용하여 계산하였다.
각-SNP 조합에 속하는 SNP들로 이루어진 모든 부울식 은 다음과 같은 순서로 생성하였다.
(1) 개의 단말노드를 가지는 모든 이진트리를 생성한 다.
(2) 이진트리의 각 내부노드에 부울 연산 AND 또는 OR 를 적용한다.
(3) 이진트리의 단말노드에 있는 각 SNP에 우성모델, 열 성모델, 우성모델의 부정(NOT), 열성모델의 부정(NOT)의 4 가지 모델을 적용한다.
우성모델이란 유전형에 우성 대립유전자가 있음으로서 표 현형이 결정되는 경우를 뜻하며, 우성모델의 부정이란 우성 대립유전자가 없음으로서 표현형이 결정되는 경우를 뜻한다.
예를 들어서, 하나의 2-SNP 조합으로 이루어지는 부울식 은 모두 32 개 이다. 2 개의 단말노드를 가지는 이진트리는 1 개 이다. 루트 노드는 AND 또는 OR 연산을 가진다. 각 단말 노드는 4 가지 모델이 가능하므로 1 * 2 * 4 * 4 = 32 개의 부울식이 가능하다.
III. 결과
임상 자료는 본 연구자 소속기관 부속병원의 천식관련 SNP 데이터를 이용하였다. 임상 자료는 총 246명에 대한 25개 SNP의 유전형(genotype)으로 이루어진다.
SNP-chip에서 얻어 전처리 과정을 거친 약 26만개 SNP 중에서 임상연구자가 과거 연구 결과와 임상 실험 정보를 이 용하여 선별한 25개의 SNP를 사용하였다.
AIA는 aspirin-induced asthma의 약자로서 아스피린 에 의해 발병한 천식을 의미하며, ATA는 aspirin-tolerant asthma의 약자로서 아스피린과 관계없는 천식을 의미한다.
본 연구에서는 AIA의 특성 SNP을 찾기 위하여 AIA를 환 자군으로 설정하고 ATA를 대조군으로 설정하여 실험하였다.
246명의 자료에서 AIA는 94명, ATA는 152명이다.
SNP ID p-value
SNP ID p-value genotype allele genotype allele SNP1 0.0268 0.1120 SNP14 0.0502 0.7867 SNP2 0.8565 0.6353 SNP15 0.9843 0.8650 SNP3 0.2178 0.6399 SNP16 0.1296 0.0986 SNP4 0.6753 0.6687 SNP17 0.7675 0.4671 SNP5 0.7069 0.9477 SNP18 0.0650 0.0252 SNP6 0.3401 0.1005 SNP19 0.2593 0.8872 SNP7 0.1881 0.0662 SNP20 0.6491 0.7025 SNP8 0.2753 0.1145 SNP21 0.9649 0.8244 SNP9 0.6175 0.4777 SNP22 0.6788 0.6706 SNP10 0.9954 0.9312 SNP23 0.0421 0.0374 SNP11 0.1760 0.0669 SNP24 0.9922 0.9041 SNP12 0.1309 0.0369 SNP25 0.5350 0.5916 SNP13 0.3515 0.9508
표 1. 각 SNP의 p-value Table 1. p-value of each SNP
표 1은 각 SNP의 p-value이다. 각 SNP의 유전형(genotype) 과 대립유전자(allele)의 p-value는 각 SNP과 환자군/대조 군의 연관성을 카이-스퀘어(chi-square) 검정으로 측정하였 다. 연관성 분석에서 유의 수준(significance level)을 0.05 로 정하면, 전체 SNP의 수가 25개 이므로 Bonferroni correction에 의하여 각 SNP의 유의 수준은 0.002 이다.
표 1에서 각 SNP은 유의 수준에 도달하지 못함을 확인 할 수 있다. 따라서 각 SNP이 단독으로 환자군/대조군과 직 접적인 연관이 있다고 말할 수 없다. 본 연구에서 제안하는 방법은 복수 SNP의 상호작용과 환자군/대조군 구분의 연관 성을 분석한다.
제 1 단계로 상호정보 값이 높은 SNP의 조합을 20위까지 선택하고 제 2 단계로 각 SNP 조합에 대하여 예측 정확도가 가장 높은 부울식을 찾는다. 부울식의 정확도는 그 부울식이 환자군과 대조군을 얼마나 정확하게 분류하는 가를 뜻한다. 2 개 SNP 조합, 3개 SNP 조합, 4개 SNP 조합, 5개 SNP 조 합의 경우를 수행하였다.
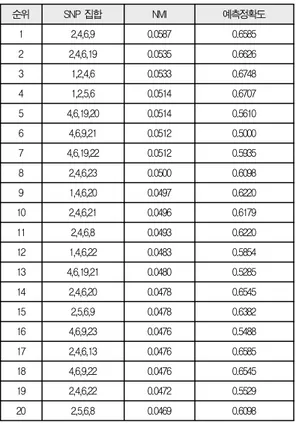
순위 SNP 집합 NMI 예측정확도
1 2,4,6,9 0.0587 0.6585
2 2,4,6,19 0.0535 0.6626
3 1,2,4,6 0.0533 0.6748
4 1,2,5,6 0.0514 0.6707
5 4,6,19,20 0.0514 0.5610
6 4,6,9,21 0.0512 0.5000
7 4,6,19,22 0.0512 0.5935
8 2,4,6,23 0.0500 0.6098
9 1,4,6,20 0.0497 0.6220
10 2,4,6,21 0.0496 0.6179
11 2,4,6,8 0.0493 0.6220
12 1,4,6,22 0.0483 0.5854
13 4,6,19,21 0.0480 0.5285
14 2,4,6,20 0.0478 0.6545
15 2,5,6,9 0.0478 0.6382
16 4,6,9,23 0.0476 0.5488
17 2,4,6,13 0.0476 0.6585
18 4,6,9,22 0.0476 0.6545
19 2,4,6,22 0.0472 0.5529
20 2,5,6,8 0.0469 0.6098
표 2. 4개 SNP 조합 중 정규상호정보 상위 20개 Table 2. NMI top 20 of 4-SNP combinations
표 2는 4개 SNP 조합 중에서 상호정보(NMI) 값이 높은 순서로 20위까지 보여준다. 표에서 “예측정확도”는 각 SNP 조합으로 이루어지는 모든 부울식 중에서 가장 높은 정확도를 뜻한다. 상호정보 값이 상위 20 안에 있는 SNP 조합 중에서 가장 높은 예측 정확도를 갖는 SNP 조합은 표에서 세 번째 항목인 {1, 2, 4, 6} 이다. 가장 높은 정규상호정보를 가지는 SNP 집합이 항상 가장 높은 예측 정확도를 나타내는 것은 아 니라는 것을 알 수 있다.
표 3은 2개 SNP 조합부터 5개 SNP 조합까지 각 경우에 가장 높은 정확도를 보이는 SNP 조합과 부울식의 정확도를 보여준다. 표 3을 보면 SNP이 4개인 경우의 부울식이 예측 정확도가 가장 높음을 알 수 있다. 이때 SNP 집합은 {1, 2, 4, 6} 이며 부울식은 식 1과 같다. 어떤 개인의 유전형 자료 (genotype data)가 식 1의 부울식을 만족하면 환자로 분류 하고 만족하지 않으면 정상으로 분류함을 뜻한다.
SNP 수
() SNP 조합
부울식의 예측정확도
2 13, 23 0.6463
3 1, 4, 6 0.6667
4 1, 2, 4, 6 0.6748
5 2, 4, 6, 9, 19 0.6545
표 3. 부울식의 예측정확도와 SNP 조합
Table 3. Boolean expression's prediction accuracy and SNP combination
SNP1은 G가 우성이고 A가 열성으로서 GG, GA, AA의 세 가지 가능한 유전형이 있다. 이 중에서 AA 유전형을 가진 사람을 aspirin-induced asthma 환자로 분류한다는 의미 이다. SNP2는 A가 우성이고 G가 열성으로서 AA, AG, GG 세 가지 유전형을 가진다. SNP4는 T가 우성이고 G가 열성 으로서 TT, TG, GG 의 유전형을 가지며, SNP6는 C가 우 성이고 T가 열성으로서 CC, CT, TT의 유전형을 가진다.
′′ ′′
′′
′′
식 1. 예측 정확도가 가장 높은 부울식 Expression 1. Best accuracy boolean expression
식 1에 나타나는 SNP1, SNP2, SNP4, SNP6 의 실제 이름은 표 4와 같다. 식 1에서 SNP1는 우성모델로 SNP2, SNP4, SNP6는 열성모델로 판정되었다.
SNP 번호 실제 SNP 명
SNP1 ALOX5_p1_1708_G>A
SNP2 B2ADR_q1_46_A>G
SNP4 CCR3_p1_520_T>G
SNP6 CysLTR1_p1_634_C>T
표 4. 식 1의 실제 SNP명
Table 4. Real SNP names of Expression 1
식 1을 통해 얻게 되는 결과는 2차원 분할표(two-way contingency table)로 표 5와 같다. 2차원 분할표의 카이- 스퀘어(chi-square) 검정값은 아래의 식에 의하여 구한다 [11].
계산된 카이스퀘어 검정값은 18.89 이며 그 확률은 0.0000138이다. 이는 식 1이 환자군/대조군을 분류하는 것 과 무관할 확률이 매우 낮음을 뜻한다.
식1의 결과
AIA ATA
임상 자료 AIA 19 75
ATA 5 147
표 5. 2차원 분할표
Table 5. two-way contingency table
각 SNP이 위치하는 유전자의 대사경로를 살펴보면 ALOX5와 CysLTR1은 류코트리엔 화합물 대사경로(leukotriene synthesis pathway) 상에 존재하며 B2ADR과 CCR3는 기 도 염증 대사경로(airway inflammation pathway) 상에 존재하는 유전자이다.
ALOX5는 한국인에서 AIA 민감성(susceptibility)이 보 고되었고[12] B2ADR와 CCR3는 천식과의 연관성이 보고 되었다[13, 14, 15]. CysLTR1은 한국인에서 AIA 발병의 유효 유전 위험 인자(significant genetic risk factor)로 보 고되었다[16].
IV. 비교 및 분석
기존의 연구 방법과 비교하고 제안하는 방법의 시간 효율 성을 분석한다.
1. MDR과의 비교
MDR(multi-dimensionality reduction)은 현재 복합 질환의 SNP 상호작용에 관한 연구에 가장 널리 쓰이는 방법 으로서, 몇 가지 복합질환 연구에 적용하여 성공적인 결과를 보였다 [3, 17, 18]. MDR은 SNP의 모든 조합과 각 조합에 포함된 SNP의 모든 유전형(genotype)에 대해서 환자군과 대조군의 비율을 계산하여, 그 비율이 정해진 기준치를 넘는 가 아닌가에 의해서 발병을 예측하는 방법이다.
MDR은 분석 결과를 룩업테이블(lookup table)의 형태 로 보여줌으로서 그 생물학적 의미를 해석하기 힘든 단점을 가지고 있다. 이에 비하여 본 연구에서 제안하는 방법은 SNP 상호작용을 부울식으로 표현함으로서, 생물학적인 의미를 유 추하기 편리하다는 장점을 가지고 있다.
제 3.2 절에서 사용한 것과 같은 임상자료에 MDR을 적 용하여 보았다.
표 6은 MDR과 상호정보+부울식(MIBA)의 비교를 나타 낸다. SNP 수에 따라 가장 높은 예측 정확도를 보이는 SNP 의 조합을 보여준다. SNP 수가 2 개인 경우는 MDR과 MIBA 가 전혀 다른 조합을 선정하였으나, SNP 수가 3, 4, 5 개인 경우에는 MDR과 MIBA가 조합을 이루는 원소가 꼭 한 개만 달라서 두 가지 방법이 유사한 결론에 도달함을 알 수 있다. 예측 정확도 면에서는 MIBA가 MDR에 비해 약간 높음을 볼 수 있다.
SNP 수
MDR MIBA
조합 정확도 조합 정확도
2 7, 22 0.5959 13, 23 0.6463
3 2, 4, 6 0.5309 1, 4, 6 0.6667
4 2, 4, 6, 9 0.6516 1, 2, 4, 6 0.6748 5 2, 4, 6, 19,
20 0.6168 2, 4, 6, 9, 19 0.6545 표 6. 임상자료에 적용한 MDR과 MIBA
Table 6. MDR and MIBA on clinical data
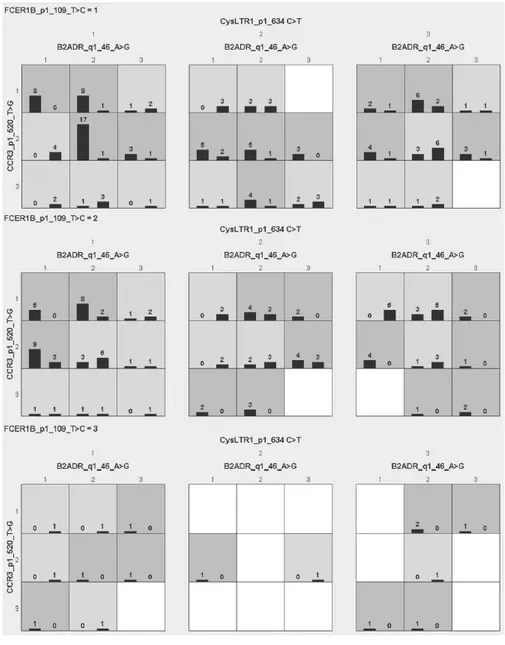
그림 2는 MDR이 선정한 4-SNP 조합을 나타내며, 그림 2에 나타나는 SNP2, SNP4, SNP6, SNP9 의 실제 이름은 표 7과 같다.
그림 2. MDR의 분석 결과 (SNP2, SNP4, SNP6, SNP9) Fig. 2. Analysis result of MDR (SNP2, SNP4, SNP6, SNP9)
SNP 번호 실제 SNP 명
SNP2 B2ADR_q1_46_A>G
SNP4 CCR3_p1_520_T>G
SNP6 CysLTR1_p1_634_C>T
SNP9 FCER1B_p1_109_T>C
표 7. 그림 2의 실제 SNP명 Table 7. Real SNP names of Fig. 2.
앞에서 언급한 바와 같이 MDR의 결과는 그림 2와 같은 룩업테이블의 형태로 표현되므로 SNP의 상호작용을 해석하 기 힘들다. 반면에 식 1과 같은 부울식은 SNP 상호작용을 간 결하게 표현하고 있다.
2. GABA 와의 비교
부울식을 이용하여 SNP의 상호작용을 표현한 기존 연구
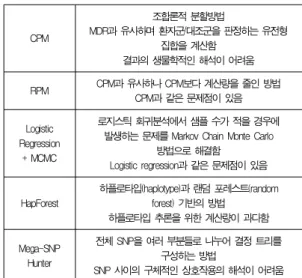
방법 특징 BEAM
베이지안(bayesian) 추론 사용 SNP간 상호작용 존재 여부를 검증할 뿐 어떤
상호작용이 있는지 설명이 어려움 Logistic
Regression
수학적으로 검증된 방법 이산적인(discrete) 유전형 데이터를 숫자로
표현하는 문제점이 있음 표 8. 기존 방법들의 특징
Table 8. Features of existing methods
CPM
조합론적 분할방법
MDR과 유사하며 환자군/대조군을 판정하는 유전형 집합을 계산함
결과의 생물학적인 해석이 어려움
RPM CPM과 유사하나 CPM보다 계산량을 줄인 방법
CPM과 같은 문제점이 있음 Logistic
Regression + MCMC
로지스틱 회귀분석에서 샘플 수가 적을 경우에 발생하는 문제를 Markov Chain Monte Carlo
방법으로 해결함 Logistic regression과 같은 문제점이 있음 HapForest
하플로타입(haplotype)과 랜덤 포레스트(random forest) 기반의 방법
하플로타입 추론을 위한 계산량이 과다함 Mega-SNP
Hunter
전체 SNP을 여러 부분들로 나누어 결정 트리를 구성하는 방법
SNP 사이의 구체적인 상호작용의 해석이 어려움 로는 GABA(genetic algorithm + boolean algebra)가
있다[19]. GABA는 유전자 알고리즘을 사용하여 부울식을 찾는다. GABA는 SNP의 수를 고정하지 않고 유전자 알고리 즘으로 하여금 최선의 SNP 수를 찾아내게 하는 유연성를 가 지고 있다.
본 연구에서 제안하는 MIBA (mutual information + boolean algebra)는 GABA에 비하여 몇 가지 장점을 가지 고 있다. 첫째 GABA에서 다루는 부울식은 괄호를 포함하고 있지 않다. 예를 들어서 (SNP1 + SNP2) * (SNP3 + SNP4)와 같은 식을 표현하지 못한다. 반면에 MIBA는 모든 부울식을 표현할 수 있다. 둘째로 GABA는 유전자 알고리즘 을 사용하므로 예측 정확도가 가장 높은 부울식을 찾아낸다는 보장이 없다. MIBA는 상호정보가 높은 SNP 조합을 가지고 모든 가능한 부울식을 다 조사하므로 예측 정확도가 가장 높 은 부울식을 찾아낼 수 있다.
최근에 SNP 상호작용 분석 연구는 50만개 이상의 SNP 을 조사하는 전체게놈 연관성 연구(genome-wide association study; GWAS)로 발전하고 있다. 최근의 연구 결과들은 수 많은 SNP 중에서 중요 SNP들을 먼저 골라내는 선택 알고리 즘을 사용하고 있다[9, 20, 21].
GABA는 전체 SNP을 대상으로 유전자 알고리즘을 사용 하므로 수많은 SNP을 다루어야 하는 GWAS로 확장하기 어 렵다. 반면에 본 연구에서 제안하는 MIBA는 중요 SNP 들을 먼저 골라내는 필터 알고리즘을 사용함으로서 GWAS로 확장 할 수 있는 가능성을 가지고 있다.
3. 기존의 다른 연구방법과의 비교
MDR은 현재 가장 널리 사용되는 SNP 연관성 연구 방법이 므로 본 연구의 결과와 비교하였으며, GABA는 SNP들의 부 울식을 사용한다는 면에서 본 연구의 내용과 유사하므로 비교 하였다. 제 1 장 서론에서 언급한 기존의 연구방법들과 본 연 구에서 제안하는 방법의 차이점을 아래의 표8에 정리하였다.
기존의 방법에 비하여 본 연구에서 제안한 방법의 주된 장점은 주어진 SNP 조합이 상호작용을 가진다는 사실에서 더 나아가 그 상호작용이 호혜적인지 배타적인지를 나타냄으 로서 그 SNP이 위치하는 유전자의 신호전달경로(signal pathway)상에서의 기능을 유추할 수 있는 단서를 제공한다 는 점이다.
4. 시간 효율성 분석
선정된 개의 SNP 조합으로 이루어지는 모든 부울식의 개수는 다음과 같다. 부울식을 나타내는 이진트리를 생각해보 자. 우선개의 단말노드(leaf node)를 가지는 이진트리의 개 수는 번째 Catalan number
이다
[22]. 각 이진트리에서 단말노드가 나타내는 각 SNP에 대해서 우성 모델(dominant model)과 열성 모델(recessive model)의 두 가지 경우를 적용하고 또한 여기에 부정(NOT)를 적용해 봄 으로써, 경우의 수가 이다. 각 이진트리의 각 내부노드 (internal node)에는 AND와 OR를 적용해 봄으로써 경우의 수
가 이다. 따라서개의 SNP 조합으로 이루어지는 모든
부울식의 개수 · 이다. 또한 부울식의 예 측 정확도를 측정하기 위해서 각 부울식을 명의 환자군/대 조군에 대해서 계산(evaluation)해야 하므로 전체 시간복잡도 는 · · 이다.
시간복잡도는 에 대해서 지수함수 이상으로 증가하지만 SNP 상호작용 연구에서 관심을 가지는 값의 범위는 매우 크지 않으므로 부울식을 생성하고 검사하는 데 걸리는 시간은
다룰 수 있는 정도이다.
상호정보가 높은 개의 SNP을 선정하는 작업은 실행시 간이 많이 필요하다. 특정한 개 SNP의 상호정보를 계산하 는 것은번의 사칙연산으로 구할 수 있으므로 문제되지 않 는다. 그러나 주어진 개의 SNP 중에서 상호정보가 높은 개의 SNP 조합을 구하는 작업에 걸리는 시간은 값이 커짐 에 따라 빠르게 증가한다.
본 연구에서 수행한 바와 같이 의학적 지식을 이용하여 선 정한 수십 개의 SNP을 대상으로 작업하는 것은 시간적으로 다룰 수 있는 정도이다. 그러나 전체게놈을 대상으로 할 경우 에는 값이 50만 이상이 되므로 실제로 계산 불가능하다. 본 연구팀은 값이 매우 큰 경우에 상호정보가 높은 개의 SNP을 선정하는 방법에 대한 연구를 진행하고 있다.
V. 결론
본 연구에서는 복합질환(complex disease)의 발병에 관 여하는 SNP들의 상호작용을 밝히기 위해서 상호정보와 부울 식을 이용하였다. 제 1 단계에서 상호정보가 높은 SNP의 조 합을 선택하고 제 2 단계에서 선택된 SNP들의 상호작용을 나타내는 부울식 중에서 발병 예측 정확도가 높은 식을 선정 하였다.
실제 임상자료를 사용하여 제안한 방법의 효율성을 검증하 였다. 현재 가장 널리 사용되는 SNP 상호작용 분석 방법인 MDR보다 다소 높은 정확도를 보였다.
기존의 SNP 상호작용 분석 방법들은 상호작용이 존재하 는 SNP의 조합을 찾아내는 데 그치고 있다. 본 연구에서는 부울식을 이용하여 구체적인 상호작용을 표현하도록 함으로 서 SNP 상호작용의 생물학적인 의미를 분석하기 용이하도록 하였다. SNP 상호작용의 생물학적인 의미를 분석하는 방법 은 최근에 시도되고 있는 다양한 개인의 질병 위험도 분석 및 진단 시스템 개발 연구에도 적용할 수 있다[23, 24].
최근에 SNP 상호작용 분석 연구는 50만개 이상의 SNP 을 조사하는 전체게놈 연관성 연구(GWAS)로 발전하고 있 다. 추후 연구과제는 제안한 방법을 개선하여 GWAS로 확장 할 수 있는 방안을 개발하는 것이다.
참고문헌
[1] A. Tarca, V. Carey, X. Chen, R. Romero, and S.
Drăghici, “Machine Learning and Its Applications
to Biology,“ PLoS Computational Biology, Vol. 3, No. 6, pp.953-963, Jun. 2007.
[2] Y. Zhang and J. Liu, "Bayesian inference of epistatic interactions in case-control studies,"
Nature Genetics, Vol. 39, No. 9, pp. 1167-1173, Aug. 2007.
[3] M. Ritchie, L. Hahn, N. Roodi, L. Bailey, W.
Dupont, F. Parl, and J. Moore, "Multifactor- dimensionality reduction reveals high-order interactions among estrogen-metabolism genes in sporadic breast cancer," American Journal of Human Genetics, Vol. 69, No. 1, pp.138-147, Jul. 2001.
[4] C. Kooperberg, I. Ruczinski, M. LeBalnc, and L.
Hsu, "Sequence analysis using logic regression,"
Genetic epidemiology, Vol. 21, pp. 626-631, 2001.
[5] M. Nelson, S. Kardia, R. Ferrell, and C. Sing, "A combinatorial partitioning method to identify multilocus genotypic partitions that predict quantitative trait variation," Genome Research, Vol. 11, pp.458-470, Jan. 2001.
[6] R. Culverhouse, T. Klevin, and W. Shannon,
"Detecting epistatic interactions contributing to quantitative traits," Genetic Epidemiology, Vol.
27, No. 2, pp. 141-152, Sept. 2004.
[7] C. Kooperberg and I. Ruczinski, "Identifying interacting SNPs using Monte Carlo logistic regression," Genetic Epidemiology, Vol. 28, No.
2, pp. 157-170, Feb. 2005.
[8] X. Chen, C. Liu, M. Zhang, and H. Zhang, "A forest-based approach to identifying gene and gene-gene interactions," Proceedings of the National Academy of Sciences of the United States of America, Vol. 104, No. 49, pp.19199- 19203, Dec. 2007.
[9] X. Wan, C. Yang, Q. Yang, H. Xue, N. Tang, and W. Yu, "MegaSNPHunter: a learning approach to detect disease prediction SNPs and high level interactions in genome wide association study,"
BMC Bioinformatics, Vol. 10, No.13, Jan. 2009.
[10] T. Cover and J. Thomas, "Elements of information
theory", 2nd ed., Wiley, 2006.
[11] 이재원, 박미라, 유한나, “생명과학 연구를 위한 통계적 방법,” 자유 아카데미, 2005.
[12] S. Kim, J. Bae, C. Suh, D. Nahm, J. Holloway, H. Park, "Polymorphism of tandem repeat in promoter of 5-lipoxygenase in ASA-intolerant asthma: a positive association with airway hyperresponsiveness," Allergy, Vol. 60, No. 6, pp.760-765, Jun. 2005.
[13] D. Contopoulos-Ioannidis, E. Manoli, and J.
Ioannidis, "Meta-analysis of the association of beta2-adrenergic receptor polymorphisms with asthma phenotypes," Journal of Allergy and Clinical Immunology, Vol. 115, No. 5, pp.
963-72, May. 2005.
[14] A. Litonjua, "The significance of beta2-adrenergic receptor polymorphisms in asthma," Current Opinion in Pulmonary Medicine, Vol. 12, No. 1, pp. 12-17, Jan. 2006.
[15] K. Fukunaga, K. Asano, X. Mao, P. Gao, M. Roberts, T. Oguma, T. Shiomi, M. Kanazawa, C. Adra, T. Shirakawa, J. Hopkin, and K. Yamaguchi,
"Genetic polymorphisms of CC chemokine receptor 3 in Japanese and British asthmatics,"
European Respiratory Journal, Vol. 17, pp.
59-46, Jan. 2001.
[16] S. Kim, J. Oh, Y. Kim, L. Palmer, C, Suh, D.
Nahm, and H. Park, "Cysteinyl leukotriene receptor 1 promoter polymorphism is associated with aspirin-intolerant asthma in males,"
Clinical & Experimental Allergy, Vol. 36, No.
4, pp. 433-439, Apr. 2006.
[17] C. Tasi, L. Lai, J. Lee, F. Chiang, J. Hwang, M. Ritchie, J. Moore, K. Hsu, C. Tseng, C.
Liau, and Y. Tseng, "Renin-angiotensin system gene polymorphisms and atrial fibrillation,"
Circulation, Vol. 109, No. 13, pp. 1640-1646, Mar. 2004.
[18] J. Moore, L. Hahn, and M. Ritchie, “Power of multifactor dimensionality reduction for detecting gene-gene interactions in the presence of genotyping error, missing data,
phenocopy, and genetic heterogeneity,” Genetic Epidemiology, Vol. 24, No. 2, pp. 150-157, Feb. 2003.
[19] K. Liang, Y. Hwang, W. Shao, E. Chen, "An algorithm for model construction and its applications to pharmacogenomic studies,"
Journal of Human Genetics, Vol.51, pp. 751–
759, Aug. 2006.
[20] J. Moore, F. Asselberg, S. Williams, "Bioinformatics challenges for genome-wide association studies,"
Bioinformatics, Vol. 26, No. 4, pp. 445-455, Jan. 2010.
[21] X. Wan, C. Yang, Q. Yang, H. X, N. Tang, W.
Yu, "Predictive rule inference for epistatic interaction detection in genome-wide association studies," Bioinformatics, Vol. 26, No. 1, pp.
30-37, Oct. 2010.
[22] T. Cormen, C. Leiserson, R. Rivest, C. Stein,
“Introduction to algorithms,” 3rd ed., MIT Press, 2009.
[23] 신진섭, 안우영, 오일용, “생체정보측정을 통한 진단시 스템 개발,” 한국컴퓨터정보학회논문지, 제 13권, 제 1 호, 219-226쪽, 2008년 1월.
[24] 김광백, 우영운, “개선된 퍼지 ART 알고리즘을 이용한 한방 자가 진단 시스템,” 한국컴퓨터정보학회논문지, 제 15권, 제 2호, 27-34쪽, 2010년 2월.
저 자 소 개 임 상 섭
2008 : 아주대학교 공학석사 2008-현재 : 아주대학교 박사과정 관심분야 : 알고리즘, 생물정보학 위 규 범
1985 : 위스컨신 대학교 이학석사 1992 : 인디애나 대학교 이학박사 1993-현재 : 아주대학교 정보컴퓨터
공학부 교수 관심분야 : 알고리즘, 생물정보학