은 주로 유전자군 GI과 GII 유전자형에 의한 것으로 알려져 있다[2].
노로바이러스 감염 진단은 위장관염 증상이 있는 사람 분변에 서 노로바이러스를 검출함으로 이루어진다[3, 4]. Enzyme-linked immunosorbent assay (ELISA), 신속항원법, 웨스턴블럿법, 역전사 중합 효소연쇄반응(reverse transcription polymerase chain reac- tion, RT-PCR)검사, 실시간 PCR (real-time PCR)검사, 라텍스응집 법, 전자현미경 등이 검출에 이용되며, 염기서열분석에 의한 노로 바이러스의 유전형 확인이 역학적 연구에 활용된다[3-6]. 이 중 현 재까지 RT-PCR 검사 또는 실시간 PCR 검사가 가장 민감도가 높은 방법으로 알려져 있다[5].
본 연구는 노로바이러스 체외진단제품 성능평가 국내 지침을 마련하기 위하여 국내외 노로바이러스 체외진단제품 허가·심사·
평가 관련 법규, 규정, 지침, 노로바이러스 제품 평가 논문 등의 자 료를 찾아 검토하고 노로바이러스 제품 관계자 설명회를 통하여 국내 각계 관계자의 의견을 수렴하여 국내실정에 적합한 노로바이 러스 체외진단제품에 대한 성능 평가 지침의 개발과정과 근거를 제시하고자 하였다.
서 론
노로바이러스는 전세계적으로 급성위장관염의 집단감염을 일 으키는 대표적인 원인 균주로, 국내에서도 겨울철마다 유행하고 있다[1]. 노로바이러스는 Caliciviridae과에 속하는 single-stranded RNA virus로, polymerase와 capsid 단백의 염기서열에 따라서 여 섯 가지(GI-GVI) 유전자군(genogroup)으로 분류된다. 이들 중 GI, GII, 그리고 GIV의 일부 균주가 사람에서 발견되었으며, 인체감염
국내 노로바이러스 체외진단제품에 대한 성능 평가 지침
Guidelines for the Performance Evaluation of In-Vitro Diagnostic Test for the Detection of Norovirus Infection in Korea
김자영1·김현수2·이선화3·오승환4·우광숙5·김성열6·민의기7
Jayoung Kim, M.D.
1, Hyun Soo Kim, M.D.
2, Sunwha Lee, M.D.
3, Seoung Hwan Oh, M.D.
4, Kwang-Sook Woo, M.D.
5, Seong-Youl Kim, Ph.D
6, Eui Kee Min, B.S.
7가톨릭관동대학교 의과대학 국제성모병원 진단검사의학과1, 한림대학교 의과대학 동탄성심병원 진단검사의학과2, 씨젠의료재단3,
인제대학교 의과대학 진단검사의학교실4, 동아대학교 의과대학 진단검사의학교실5, (주)씨젠 학술 지원실6, (주)에스디7
Department of Laboratory Medicine
1, International St.Mary’s hospital, Catholic Kwandong University College of Medicine, Incheon; Department of Laboratory Medicine
2, Dongtan Sacred Heart Hospital, Hallym University College of Medicine, Hwaseong; Seegene Medical Foundation
3, Seoul; Department of Laboratory Medicine
4, Inje University College of Medicine, Busan; Department of Laboratory Medicine
5, Dong-A University College of Medicine, Busan; Department of Academic Support Affair
6, Seegene Inc., Seoul; Standardia Diagnostics, Inc.
7, Yongin, Korea
https://doi.org/10.3343/lmo.2017.7.1.1
Corresponding author: Hyun Soo Kim
Department of Laboratory Medicine, Hallym University Dongtan Sacred Heart Hospital, Hallym University College of Medicine, 7 Keunjaebong-gil, Hwaseong 18450, Korea
Tel: +82-31-8086-2775, Fax: +82-31-8086-2789, E-mail: [email protected] Received: January 19, 2016
Revision received: March 3, 2016 Accepted: March 4, 2016
This article is available from http://www.labmedonline.org 2017, Laboratory Medicine Online
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Norovirus is a leading cause of epidemic and sporadic acute gastroenteritis worldwide. Rapid and accurate detection of norovirus is essential for the prevention and control of norovirus outbreaks. The purpose of this study was to propose and develop a process for establishing appropriate standardized guidelines for the approval and evaluation of in vitro diagnostic medical devices (IVDD) for norovirus detection in Korea based on the related laws, regulations, and guidelines of USA, Europe, and Korea. We expect that this study could be used for diagnostic test standardization and the approval and evaluation of domestic norovirus diagnostic devices. We also expect the results will contribute to industrial expansion and public health promotion.
Key Words: Norovirus, In-Vitro Diagnostic Device, Evaluation, Guideline
대상 및 방법
1. 문헌조사
1) 국내 식품의약품안전처 체외진단제품 관련 법규, 규정, 지침 조사
2015년도 의료기기법 시행규칙 제5조(제조허가의 절차), 제6조 (제조인증의 절차), 제9조(기술문서 등의 심사), 제15조(수입업허가 등) 및 제30조(수입허가 신청 등) [7]와 의료기기법 허가·신고·심사 등에 관한 규정, 의료기기 품목 및 품목별 등급에 관한 규정 및 의 료기기의 안정성시험 기준을 조사하였다[8-11]. 또한, 2015년까지 식품의약품안전처(식약처)에서 발간된 유사 지침인 “로타바이러 스 및 아데노바이러스의 허가심사 가이드라인”, “산전검사 중 거대 세포바이러스(CMV) 체외진단용 의료기기 허가심사 가이드라인”,
“산전검사 중 풍진바이러스(Rubella virus) 체외진단용 의료기기 허가심사 가이드라인” 및 “산전검사 중 단순포진바이러스(HSV) 체외진단용 의료기기 허가심사 가이드라인”을 조사하였다[12-14].
2) 미국과 유럽 체외진단제품 허가 심사 평가 관련 법규, 규정, 지침 조사
미국 FDA (U.S. Food and Drug Administration)의 체외진단제품 허가·심사·평가 체계를 조사하였으며 미국 FDA OIR (Office of In Vitro Diagnostic and Radiological Health) (formerly OVID) 지침 중 노로바이러스 혈청학적 시약 및 다중분석 PCR 등에 대한 특별 관리(Special control) 규정 및 FDA에 제출된 노로바이러스 제품에 대한 관련 심사서류인 시판전신고(Premarket notification, 510(k)) 를 조사하였다[15-18]. 또한, 미국 민간 표준 연구소(Clinical Labo- ratory Standard Institute, CLSI)의 체외진단분석기용 시약 관련 지 침 문서를 조사 및 분석하였다[19, 20]. 유럽의 경우 CE (Conformité Européenne [European Conformity]) marker 획득을 위해 In Vitro Diagnostic Medical Device Directive, IVDMDD (98/79/EC)에 규 정된 체외진단제품 허가·심사·평가 체계를 조사하고, 유럽 체외진 단제품 관련문서 중에 노로바이러스 제품에 대한 지침이 없으므로
체외진단제품 허가 관련문서를 조사하였다[21, 22].
3) 노로바이러스 진단제품 평가 논문 조사
노로바이러스 진단제품의 평가 논문을 PubMed에서 찾아 평가 항목을 조사하였다. PubMed 분석용어는 “Evaluation + Norovi- rus”이고 2010년 이후 내용을 조사하였다[3-6].
2. 관련자 회의 및 설명회를 통한 의견 수렴
노로바이러스 체외진단제품 허가·심사·평가 지침 제안을 위해 진단검사의학회, 노로바이러스 시약 수입업체, 제조업체 관련자 등 으로 전문가협의체를 구성하여 지침을 작성하였고 설명회를 통해 각 계 관련자의 의견을 수렴하였다.
결 과
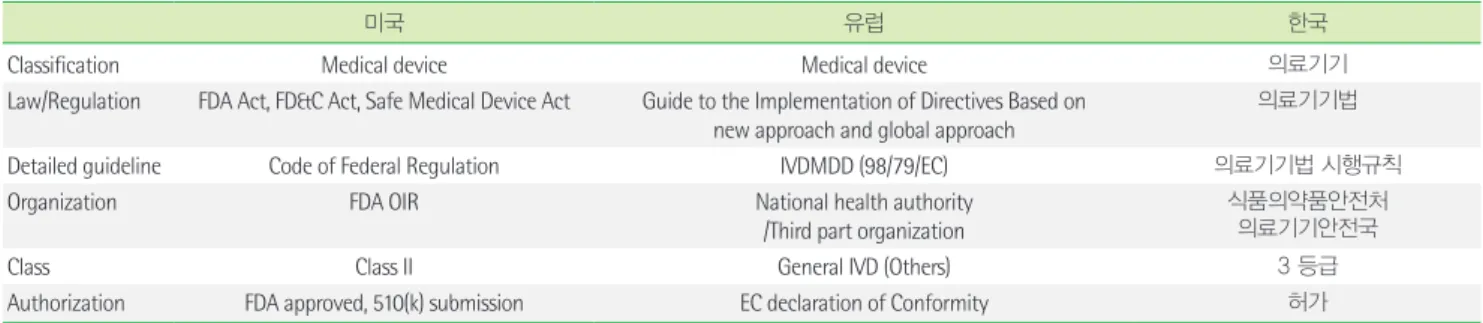
1. 국내외 노로바이러스 제품 관련 법규 및 규정 조사 국내외 노로바이러스 제품 관련 법규를 요약하여 Table 1에 기 술하였다.
1) 국내 식품의약품안전처 법규 및 규정 조사
식약처 규정에 따르면 노로바이러스 체외진단제품은 3등급 제 품으로 분류되어 ‘의료기기 제조(수입) 허가신청서’와 ‘의료기기 기술문서 등 심사의뢰서’를 식약처 장에게 제출하고 허가 받아야 한다. 허가신청서 기재 항목은 명칭(제품명, 품목명, 모델명), 분류 번호(등급), 모양 및 구조, 원재료, 제조방법, 사용목적, 사용방법, 사용 시 주의사항, 포장단위, 저장방법 및 사용기간, 시험규격, 제 조원(수입 또는 제조공정 전부 위탁의 경우), 허가(인증)조건, 비고 순이다[7-11].
2) 미국과 유럽 법규, 규정, 지침
미국은 Class II로 분류되어 FDA 시판전 승인이 필요하고 특별 관리(special control)가 적용되어 특별 라벨링 규정, 성능 기준과
Table 1. Product classification of norovirus detection devices
미국 유럽 한국
Classification Medical device Medical device 의료기기
Law/Regulation FDA Act, FD&C Act, Safe Medical Device Act Guide to the Implementation of Directives Based on new approach and global approach
의료기기법
Detailed guideline Code of Federal Regulation IVDMDD (98/79/EC) 의료기기법 시행규칙
Organization FDA OIR National health authority
/Third part organization
식품의약품안전처 의료기기안전국
Class Class II General IVD (Others) 3 등급
Authorization FDA approved, 510(k) submission EC declaration of Conformity 허가
Abberations: OIR, the Office of In Vitro Diagnostics and Radiological Health; PMN, Premarket Notification; IVDMDD, In Vitro Diagnostic Medical Device Directive.
사후 감시를 포함한다[15-17]. 판매자는 510(k)에 제외되지 않는 한, 판매 전 90일 이내에 FDA 에 510(k) 서류를 접수해야 한다[15-18].
관련 규정으로 미국 FDA OIR의 “Class II 특별규제 지침서: 노로바 이러스 혈청학적 시약들”이 있고 “Class II 특별규제: 인체 분변검 체에서 미생물과 독소유전자 검출 및 동정을 위한 장관미생물 다 중핵산분석 기반 검사” 일부에 노로바이러스 GI/GII 유전자형 검 출 시약 내용이 포함되어 있다. 제품 설명시 주시약 이외에 보조시 약, 각 정도관리물질(양성, 음성 및 내부정도 관리물질) 및 관련 소 프트웨어에 대한 정의와 평가도 포함하고 노로바이러스 혈청학적 시약 지침은 시판후 검증을 포함한다[16, 17]. 미국 CLSI 문서는 감 염질환 검사법에 대한 설계(design), 개발(development), 검증(vali- dation) 단계의 성능평가에 관한 제조사와 사용자 지침, 검사법 수 행 (implementation) 단계의 성능평가에 관한 지침 및 허가심사를 위한 검증 단계에 적용되는 성능평가 지침을 제공한다. 그러나 노 로바이러스에 대한 개별 지침은 개발되지 않았고 감염질환의 분자 진단 검사 지침(CLSI MM03-Ed3) 과 장관 내 미생물의 다중핵산측 정법 검사 지침(CLSI MM17-A) 내에 포함되어 있다[19, 20]. 한편, 유 럽의 경우 풍진, 톡소플라스모시스, 거대세포바이러스, 클라미디 아를 제외한 바이러스 체외진단제품은 General IVD (Others)에 속하여 자가적합성 선언(EC declaration of Conformity)만으로 시 판이 가능하다. Others제품은 지정된 유럽대리인을 통해 Compe- tent Authority에 기술문서를 제출하며 승인 이후 제조업자의 자가 선언으로 CE 마크를 부착할 수 있다. 기술문서에는 일반적인 서술, 디자인 정보, 제조사, 위험 분석 및 위험 관리 파일, 성능 평가 데이 터, 라벨 및 IFU 정보, 안정성 연구, 질관리 및 질 보증에 대한 서류 및 필요 물품에 대한 체크리스트를 포함한다[21, 22].
3) 국내 노로바이러스 체외진단기기 성능 평가 지침
본 지침은 개요, 용어의 정의, 관련 규정, 신청서 기재 항목 및 기 술문서 제출자료, 제조. 수입허가 신청서 기재항목, 기술문서 등의 심사를 위한 제출자료, 성능시험에 대한 지침, 참고문헌으로 구성 하였다. 사용목적에서 해당 검체로 분변검체를 명시하고, 임상적 적용분야에 대한 기술을 권장하였다. 또한, 시험 항목별로 필요한 정도관리 질의 범위를 고, 중, 저 역가를 포함한 최소 3개 이상으로 정하고 이에 대한 사용도 권장하였다.
2. 문헌조사 내용을 근거로 각 성능평가 항목의 구체적 방법 결정
1) 분석적 민감도(검출한계)
검출한계 측정을 위한 미국 FDA 노로바이러스 혈청학적 시약 및 유전형 검사 지침에서는 특성이 입증된 최소 하나 이상의 GI과 GII (GII.4 권장) 유전자형 양성 분변검체를 단계희석하여 측정한
다. 대략적인 검출한계(limit of detection)는 각 희석 농도의 검체 를 3회 반복측정하여 최소 2번이 양성으로 나오는 노로바이러스 수준(예: 바이러스 입자 수/분변 gram, RNA copy 수/분변 gram)으 로 추정한다. 최소검출한계는 추정된 최소검출한계 값을 가진 검 체를 최소 20회 반복측정하여 반복측정의 95% 이상 양성 결과를 나타내는 최소 바이러스 농도로 결정한다. 분변검체에서 노로바이 러스 입자수는 전자현미경을 이용하여 측정하고(입자 수/분변 gram), RNA copy 수는 기존의 성능이 입증된 실시간 PCR검사로 측정한 다(RNA copy 수/분변 gram) [16, 17]. CLSI MM17-A의 검출한계는 CLSI EP17를 따라 최소 60회 반복측정하여 58번이 양성(최저검출 농도에서 5% 이상의 위음성 결과를 산출하지 않는 핵산 농도의 95% 신뢰구간)이면서 blank와 유의한 차이를 보이는 핵산 농도로 정한다[19, 20]. 본 지침의 검출한계 설정에 사용된 검체 종류, 바이 러스 유전형 및 반복측정 횟수는 FDA 지침을 따르고 측정계산법 은 RNA copy 수(RNA copy 수/분변 gram)를 제시하였다.
2) 유전자형별 반응도(Strain reactivity)
미국 FDA 노로바이러스 혈청학적 형별검사에 따르면 GI 8종(GI.1, GI.2, GI.3, GI.4, GI.5, GI.6, GI.7, GI.8), GII 16종(GII.1-10, GII.12, GII.13, GII.14, GII.15, GII.16, GII.17) 및 GIV 1종(GIV.1) 양성 분변 검체를 포함하도록 추천하고 강양성(고농도)과 판정 기준치 근처 값을 갖는 저농도 양성검체 이용도 권장한다[16]. 본 지침에서는 한 종류 이상의 GI형과 GII.2, GII.3, GII.4, GII.6 및 GII.13형 등을 포 함한 양성 분변검체와 각 유전자형별 고농도 양성(high positive) 및 저농도 양성(low positive) 분변검체를 이용하도록 권장하였다.
3) 분석적 특이도(간섭)
간섭 평가를 위한 미국 FDA 노로바이러스 혈청학적 검사 및 유 전자 검사 지침에서는 권장물질로 혈액, 뮤신, 변비나 설사를 유발 하는 흔한 약제, 항생제, 진통제, 조영제(barium sulfate) 등 최소 11 종 이상을 제시하고 있다. 가능한 가장 임상적으로 의미있는 농도 에서 각 간섭물질을 평가하며 판정기준치 근처 값을 갖는 각각 최 소 3개의 양성 분변검체(약양성검체) 및 음성 분변검체를 사용한 다[16, 17]. CLSI MM03-Ed3에서는 염기서열 오염물의 검출 및 기타 가능한 간섭효과 평가로 20-30개의 음성 blank 검체를 반복측정 한다. 이때 양성결과가 있어서는 안되며, 음성기저신호(혹은 다른 반응측정)가 증가되거나 예측된 것과 달라서는 안된다[19]. CLSI MM17-A에서는 간섭물질의 평가 시 임상적 상황이 발생할 수 있는 최대 농도로 실시할 것을 규정하고 있다[20]. 본 지침에서도 FDA 노로바이러스 혈청학적 시약 관련 규정을 참조하여 간섭물질을 첨 가한 최소 3개의 약양성 분변검체와 3개의 음성 분변검체를 3회 이상 반복측정하여 간섭물질을 함유한 검체(test sample)와 함유
하지 않은 검체(control sample)의 결과값을 제시하고 두 값의 차 이가 허용범위(dmax, 지정한 조건에 맞는 데이터베이스의 필드 값 중 가장 큰 수)보다 작은지 판단하도록 하였다.
4) 분석적 특이도(교차반응)
교차반응 평가를 위한 미국 FDA 노로바이러스 혈청학적 검사 및 유전자 검사 지침에서는 위장관염 증상을 가진 환자 분변에서 검출 가능한 50종 이상의 다양한 세균들과 바이러스에 대한 평가 를 권장한다. 분변검체 내 세균 농도가 106 CFU/mL 이상, 바이러스 농도가105 PFU/mL 이상에서 시행하며[16, 17], 510(k)에서는 바이 러스 농도 기준으로 105 TCID50/mL 이상도 사용한다[18]. 이에 본 지침에서는 분변검체 내 세균 농도 기준으로 106 CFU/mL 이상, 바 이러스 농도 기준으로 105 PFU/mL와 105 TCID50/mL 이상을 모두 포함하였다.
5) 정밀도(반복, 재현성)
정밀도 평가를 위한 미국 FDA 노로바이러스 관련 혈청학적 검 사 및 유전자 검사 지침에서는 임상적으로 중요한 농도 범위를 포 함하는 4-6개의 검체들을 최소 12일간(연속적일 필요 없음) 매 검 사 차수(run) 마다 각 검체 당 3번 반복 측정한다. 재현성 평가는 최소 3곳 이상의 기관(외부검사실 2곳과 당해 검사실 혹은 외부검 사실 3곳)에서 매일 최소 2인의 검사자가 각각의 검사실에서 최소 5일 동안, 매일 최소 2회 이상, 매 검사마다 3회 반복 측정한다. 이 때 최소 2-3가지의 노로바이러스 아형(GII.4 포함)을 포함하고 이 때 판정기준치 근처를 포함한 최소 3가지 농도를 가진 분변검체들 (음성검체, 약양성검체 및 중등양성 검체)로 구성할 것을 권장하고 제조사 포함 3곳 이상에서 평가하도록 한다[16, 17]. CLSI MM03-Ed 의 유전자 검출에 대한 정량검사는 10-20일간 시행한 정도관리결 과로 표준편차와 변이계수를 계산하는 CLSI EP05 정밀도 평가를 따른다[19]. 본 지침에서는 기존 국내 유사지침과 동일하게 3가지 농도 이상의 분변검체들로 최소 10일 동안(20일 이상 권장), 1일 1 회 이상, 2-3번 이상 반복측정(반복성)하고 로트간 정밀도(lot-to- lot precision)는 최소 3종 이상의 로트를 사용하며 2 곳 이상의 기 관에서 각 장소마다 2인 이상의 검사자가 최소 5일 동안, 최소 1일 2회 이상, 2번 이상 반복 측정(재현성)할 것을 권장한다.
6) 임상적 성능 평가(임상민감도, 임상특이도)
임상적 성능 평가를 위한 미국 FDA 노로바이러스 관련 혈청학 적 검사 및 유전자 검사 지침에서는 시약 사용 목적으로 급성위장 관염 환자의 노로바이러스 진단인지 역학조사인지로 구분하여 명 시하도록 한다. 전향적으로 수집한 증상 발현 3일 이내의 신선 분 변검체가 더 좋다. 보관 분변검체도 가능하나 선택적으로 사용하
면 안된다. 신선검체와 보관검체를 같이 평가했다면 두 군으로 나 누어 분석하기를 권장한다. 임상시험 장소는 시약이 사용될 각기 다른 곳에 위치한 최소 3곳 이상의 기관이며 그 중 최소 2곳은 미 국 내에 있어야 한다[16, 17]. 임상 시험시 참고방법에 의한 임상적 진단 참값(clinical diagnostic truth) 판정은 실시간 RT-PCR 검사 결 과와 상관없이 염기서열 분석을 수반한 통상적인(conventional) PCR 검사의 노로바이러스 검출로 판정한다[16]. 본 지침에서도 분 변검체 수집 방법, 선정기준, 추천 검체, 권장 통계방법 및 임상적 진단 참값(clinical diagnostic truth) 판정 기준은 FDA 지침과 동일 하게 제시하였다. 그러나, 사용목적을 개별 환자 진단 혹은 역학조 사 간에 구분하도록 규정하지 않았고, 임상시험 기관은 최소 2곳 으로 하였다.
7) 상관성
상관성 평가를 위한 미국 FDA 노로바이러스 혈청학적 검사의 참고검사법은 FDA 인정 혹은 승인 받은 실시간 RT-PCR 검사이나 양방향 염기서열 분석을 수반한 통상적인 PCR 검사이다[16]. 유전 자 검출시약은 통상 1,500개 이상의 전향적 수집 분변검체로 규정 하고 다음 세 가지 검사를 참고검사법으로 활용한다. 1) FDA 승인 기기, 2) 양방향 염기서열 분석을 수반한 검증된 PCR 검사 2종, 3) 양방향 염기서열 분석을 수반한 검증된 PCR 검사 1종과 배양법 혹은 양방향 염기서열 분석을 수반한 검증된 PCR 검사 1종과 FDA 승인된 혈청학적 검사 제품[17]. 본 지침에서도 기존 국내 유사 지 침을 참고하여 해당 제품과 측정 원리와 항목이 가장 유사한, 가 능하면 2종의 국내 허가 제품 또는 외국 허가 제품(국내 허가 제품 이 없을 시)과의 비교시험을 시행하고 정량 검사는 최소 2번 이상 반복측정을 권장하였다.
고 찰
현재까지 개발된 노로바이러스 체외진단제품들의 허용검체는 분변검체이다[3, 4]. 노로바이러스 체외진단제품의 분류 등급은 국 가마다 약간씩 다른데, 미국은 Class II로 시판전 승인이 필요하며 유럽은 General IVD (Others)에 속해 자가 선언만으로 시판 가능 하다[16, 17, 21, 22]. 한국은 3 등급 제품으로 식약처 허가가 필수적 이다[8, 9]. 미국 및 유럽의 지침과 비교한 본 지침의 구성상 특징은 다음과 같다. 첫째, 혈청학적 검사법과 유전자 검출법이 각자 독립 된 지침을 가지는 미국 FDA 지침 및 CLSI문서[16, 17, 19, 20]와 달 리 본 지침 내에는 이 두 가지 검사법을 같이 포함하였다. 둘째, “진 단검사의학 용어집”[23]에 따른 정확한 용어 정의를 지침 내에 제 공하여 이를 통해 혼란을 최소화하고 검사를 정확히 이해하여 학 계뿐 만 아니라 체외진단제품을 직접 생산하는 산업계 및 수입 판
매사 등 다양한 기관들 간의 소통을 원활하게 하는 데 도움을 주 고자 하였다. 셋째는 지침 내에 국내 제조된 노로바이러스 체외 진 단제품의 구체적인 기술 예시를 제시함으로써 실제 제출서류 작성 시 활용할 수 있도록 하였다. 넷째, 기존 유사 지침에는 없었지만, 본 지침에서는 미국FDA 지침과 CLSI 문서[16-20]를 참조하여 임상 적 적용분야에 대한 기술과 정도관리물질 범위에 대한 구체적인 기술을 권장사항으로 포함하였다. 그러나 미국 지침과는 달리 본 지침에는 보조시약 평가법, 각 정도관리물질의 세분화된 정의 및 평가법, 관련 소프트 웨어에 대한 정의와 유전자 발현 분석에 대한 평가방법에 대한 내용[16-20]은 포함하지 않았다. 또한, 미국의 노 로바이러스 체외진단제품 관련 지침들은 새로운 유전자형 변이형 검출을 위한 주기적인(매년 권장) 시판후 검정을 포함하고 있으나 [16], 현재 국내 제품을 대상으로는 어떤 노로바이러스 유전자형을 검출하는지에 대한 검증이 확립되지 않았고, 매년 유행형에 대한 추적관찰 및 시약의 갱신을 지속적으로 실행하고 있지 못하는 국 내 실정을 고려하여 본 지침에는 시판 후 검정을 포함하지 않았다.
체외진단기기의 성능 평가 시에는 실제 다양한 항목에 대한 성 능평가가 필요하다. 본 지침에서는 기존 국내 규정 및 2015년까지 식약처에서 발간된 유사 지침에서 제시한 분석적 민감도(검출한 계), 분석적 특이도(간섭, 교차반응), 정밀도(반복, 재현성), 임상적 성능 평가(임상적 민감도, 임상적 특이도) 및 상관성 평가에 대한 평가 기준[7-11]을 참고하였다. 한편, 기존 국내 유사 지침에는 “정 확도” 평가 항목이 있고 표준품, 상용패널, 배양액 등을 이용하여 평가하도록 하고 있다[12-14]. 그러나, 현재 미국 FDA 지침이나 CLSI 문서 및 유럽 지침에서는 감염질환 진단시약에 대해 “정확도(accu- racy)” 평가항목은 없다[15-22], 다만, 분석적 반응도 평가로 유전자 형별 반응도 평가가 포함되는데, 미국 FDA 노로바이러스 혈청학적 지침에서는 총 25종 유전자형를 추천하고 있다[16]. 이에 본 지침에 서도 정확도 항목 대신 국내 검출율이 높은 최소 6종(한 종류 이 상의 GI형을 포함한 GII.2, GII.3, GII.4, GII.6 및 GII.13형 등) [2-6]
에 대한 유전자형 반응도 평가를 포함하였다. 한편, 미국 FDA 지침 에서는 평가 권장사항인 총 11종 이상의 간섭물질 및 50종 이상의 교차반응 물질[16]을 단지 예시로 제시하였으며, 미국 510(k) [18]를 참고하여 교차반응 평가시의 바이러스 농도 기준에 105 TCID50/mL 이상을 병기하였다. 반복성 및 재현성 평가는 기존 국내 유사지침 을 참조하였다. 임상적 성능평가 시 선호 참고방법, 선호 검체, 불일 치 결과시 원인분석 검사법 등에 대해서는 미국 FDA 지침[16, 17]
과 동일하지만 이를 권장사항으로 포함하고 그 외 임상 시험에 관 한 사항은 기존 국내 유사지침과 동일하게 구성하였다.
현재 국내외 많은 노로바이러스 항원과 유전자를 검출하는 시 약이 개발되어 사용되고 있으며 향후에는 노로바이러스뿐 아니라 다른 급성설사 원인 바이러스, 세균 및 기생충까지를 한꺼번에 진
단하는 다중 분자진단 제품이 더 많이 개발될 것으로 보인다. 본 연구에서는 국내 실정에 맞는 노로바이러스 체외진단제품의 허가·
심사·평가 지침에 대한 개발과정 근거를 제시하고 적절한 표준화 지침을 확립하고자 하였다. 이를 통해 투명한 심사 평가 체계를 제 공하고 검사표준화에 기여함으로써 국산제품의 품질에 대한 국제 신뢰성 제고, 해외 허가 촉진 등을 통한 국가경쟁력 제고와 정확한 검사를 통한 국민 건강의 질적 향상에 기여할 수 있을 것으로 생 각한다.
요 약
노로바이러스는 전세계적으로 급성위장관염을 일으키는 주요 원인균이다. 노로바이러스는 전염성이 강한 바이러스이므로 노로 바이러스감염의 유행을 예방하기 위해서는 조기진단이 매우 중요 하다. 본 연구는 노로바이러스 체외진단제품을 대상으로 국내 및 미국, 유럽을 포함한 법률, 규정 및 지침을 바탕으로 국내실정에 적 합한 노로바이러스 체외진단제품에 대한 표준화된 허가 및 성능 평가 지침의 개발과정과 지침의 근거를 제시하고자 하였다. 본 연 구 의 결과는 검사 표준화와 국내 노로바이러스 체외진단제품들 의 허가·심사·평가에 유용하게 활용되고 관련 산업계 및 국민 보 건 증진에 기여할 수 있을 것이다.
감사의 글
본 연구는 2015년 식품의약품안전처의 연구개발비(15172MFDS433) 로 수행되었으며 이에 감사 드립니다.
REFERENCES
1. Park DJ, Kim JS, Park JY, Kim HS, Song W, Kim HS, et al. [Epidemio- logical analysis of norovirus infection between March 2007 and Febru- ary 2010]. Korean J Lab Med 2010;30:647-53.
2. Desselberger U, Goodfellow I. Noroviruses: a global cause of acute gastroenteritis. Lancet Infect Dis 2014;14:664-5.
3. Lee H, Park Y, Kim M, Jee Y, Cheon DS, Jeong HS, et al. Development of a latex agglutination test for norovirus detection. J Microbiol 2010;
48:419-25.
4. Kim HS, Hyun J, Kim JS, Song W, Kang HJ, Lee KM. Evaluation of the SD Bioline Norovirus rapid immunochromatography test using fecal specimens from Korean gastroenteritis patients. J Virol Methods 2012;
186:94-8.
5. Hyun J, Kim HS, Kim HS, Lee KM. Evaluation of a new real-time re-
verse transcription polymerase chain reaction assay for detection of norovirus in fecal specimens. Diagn Microbiol Infect Dis 2014;78:40-4.
6. Vinje J. Advances in laboratory methods for detection and typing of norovirus. J Clin Microbiol 2015;53:373-81.
7. Medical Device Technique Law No. 13116 (2015. 4), Medical Device Technology Enforcement Decree No. 24480 (2015.7), Medical Device Technology Enforcement Rule No. 1181 (2015.7).
8. Regulations on medical device licensing, notification and examina- tion Article 33 (Types and Requirements of Examination Documents of Medical Devices for In-vitro Diagnosis) (2015.2) Ministry of Food and Drug Safety.
9. Regulations on the Classification of Medical Device. Safety Notifica- tion No. 2015-18 (May 4, 2015), Ministry of Food and Drug Safety.
10. Clinical performance test management guideline of in vitro diagnostic medical device Article 24 (2015.7).
11. In vitro diagnostic medical devices Guidelines for approval and screening of rotavirus and adenovirus (2015.2) Ministry of Food and Drug Safety.
12. Guideline for screening gynecological virus (CMV) medical devices for in-vitro diagnostics during prenatal testing (2015.2) Ministry of Food and Drug Safety.
13. Rubella virus in prenatal test Guidelines for screening for medical de- vices for in vitro diagnostics (2015.2) Ministry of Food and Drug Safety.
14. Guidelines for Screening of Medical Devices for In Vitro Diagnosis of Herpes Simplex Virus (HSV) during Prenatal Examination (2015.2) Ministry of Food and Drug Safety.
15. U.S. Food and Drug Administration (FDA). Guidance for Industry and Food and Drug Administration Staff- Statistical guidance on reporting results from studies evaluating diagnostic tests, http://www.fda.gov/
RegulatoryInformation /Guidances/ucm071148.htm (Updated on March 13, 2007).
16. U.S. FDA. Guidance for Industry and Food and Drug Administration
Staff-Class II Special Controls Guidance Document: Norovirus Sero- logical Reagents, http://www.fda.gov/MedicalDevices/DeviceRegula- tionandGuidance/Guidancedocuments/ucm295088.htm (Updated on March 9, 2012).
17. U.S. FDA. Guidance for Industry and Food and Drug Administration Staff- Class II Special Controls Guideline: Gastrointestinal Microorgan- ism Multiplex Nucleic Acid-Based Assays for Detection and Identifica- tion of Microorganisms and Toxin Genes from Human Stool Specimens.
http://www.fda.gov/downloads/MedicalDevices/DeviceRegulation- andGuidance/GuidanceDocuments/UCM470559.pdf (Updated on No- vember 2, 2015).
18. U.S. FDA. 510(k) premarket notification. https://www.accessdata.fda.
gov/scripts/cdrh/cfdocs/cfpmn/pmn.cfm (Updated on January 2016).
19. Clinical and Laboratory Standards Institute. Molecular Diagnostic Meth- ods for Infectious Disease, 3rd Edition, MM03-Ed3, Wayne, PA: Clini- cal and Laboratory Standards Institute, 2015.
20. Clinical and Laboratory Standards Institute. Verification and validation of multiplex nucleic acid assays; approved guideline, MM17-A, Wayne, PA: Clinical and Laboratory Standards Institute, 2008.
21. European Community. Directive 98/79/EC of the European Parliament and of the Council of 27 October 1998 on in vitro diagnostic medical devices. http://ec.europa.eu/growth/single-market/european-standards/
harmonised-standards/iv-diagnostic-medical-devices/index_en.htm (Updated on January 2016).
22. European Commission. 2009/886/EC: Commission Decision of 27 No- vember 2009 amending Decision 2002/364/EC on common technical specifications for in vitro diagnostic medical devices (notified under document C (2009) http://www.itczlin.cz/editor/files/root_f/cz-MDD/
DIRECTIVES/Corr_2009_886_EC. pdf (Updated on January 2016).
23. Korean Society for Laboratory Medicine. Medical glossary for labora- tory medicine. Academia, 2010.