Journal of the Korean Chemical Society 2020, Vol. 64, No. 4
Printed in the Republic of Korea https://doi.org/10.5012/jkcs.2020.64.4.239
-239-
Note
New Synthesis of Thioflavones by the Regioselective Cyclization of 1-(2-Benz- ylthio)phenyl-3-phenylprop-2-yn-1-ones Using Hydrobromic Acid
Jae In Lee
Department of Chemistry, College of Science and Technology, Duksung Women’s University, Seoul 01369, Korea.
*E-mail: [email protected]
(Received April 10, 2020; Accepted May 8, 2020) Key words: Thioflavones, Condensation, Acylation, 6-Endo cyclization
Thioflavones are thio analogs of naturally occurring fla- vones and have the skeleton of a thiochroman-4-one ring with 2-substituted aryl groups. They exhibit a variety of pharmacological activities, such as antibacterial and anti- fungal effects,1 antiviral activity2 against enterovirus and coxsackievirus, and antiproliferative activity3 on human breast cancer. They also display inhibitory action against tumor cells4 and induced endotherium-dependent vaso- relaxation through activation of the epidermal growth fac- tor (EGF) receptor.5
Several types of reactions for the synthesis of thiofla- vones have been described.6 In general, thioflavones were synthesized by the cyclic condensation of thiophenols with ethyl benzoylacetates using polyphosphoric acid at 90 oC in low yields (Scheme 1(a)).7 The condensation of methyl 2-mercaptobenzoate with N-benzoylhydrazones with an excess of LDA gave the C-acylated intermediates, which underwent subsequent addition and hydrolysis with 3 N HCl under reflux to produce thioflavones (Scheme 1(b)).8 The condensation of 2’-haloacetophenones and methyl phenyldithioesters using a 1.5 equiv. of sodium hydride in DMF also afforded thioflavones in low to moderate yields (Scheme 1(c)).9 The cyclodehydration of 1-(2-mercaptophe- nyl)-3-phenylpropan-1,3-diones, which were prepared from 2’-mercaptoacetophenone and N-methoxy-N-methyl ben- zamides using sulfuric acid in CH3CN afforded the thio- flavones in high yields (Scheme 1(d)).10
Alternatively, the intramolecular Friedel-Crafts acylation of 3-(arylthio)-3-phenylpropanoic acids, which were derived from the 1,4-addition of thiophenols to trans-cinnamic acid using sulfuric acid, afforded thioflavanones, which were dehydrogenated further with DDQ in refluxing benzene to give the thioflavones in moderate yields (Scheme 1(e)).11 The reaction of S-aroyl derivatives of 2-mercaptobenzoic acid with N-phenyl(triphenylphosphoranylidene)ethenimine
or (trimethylsilyl)methylenetriphenylphosphorane produced the corresponding acylphosphoranes, which underwent Wit- tig type cyclization in refluxing THF to give thioflavones (Scheme 1(f)).12
On the other hand, tandem reactions using chalcone or alkynone intermediates allowed the convenient synthesis of thioflavones. The treatment of 3-aryl-1-(2-tert-butylthio) phenylprop-2-en-1-ones, which had been prepared from the condensation of arylaldehydes with 2’-(tert-butylthio)ace-
Scheme 1. Several methods for the synthesis of thioflavones.
Journal of the Korean Chemical Society
240 Jae In Lee
tophenones using NaOH, with 3 equiv. of iodine in the presence of NaHCO3 in refluxing EtCN afforded the thio- flavones (Scheme 2(a)).13 The reaction of 2’-iodochalcones and potassium O-ethyl dithiocarbonate as a sulfur source in the presence of 10 mol% Cu(OAc)2 afforded the thio- flavanones in situ, which then underwent sequential oxi- dation with sulfuric acid at 80 oC to give the thioflavones (Scheme 2(b)).14 Tandem substitution-SNAr reaction of β- chlorochalcones, which had been prepared by the iron- catalyzed addition of aryl chlorides to arylacetylene, with sodium hydrosulfide in the presence of 2 equiv. of Cs2CO3 in DMSO at 140oC afforded thioflavones (Scheme 2(c)).15 The addition of sodium hydrosulfide16 or sodium sulfide17 to 2’-bromoalkynones produced the thiolate adducts rap- idly, which then underwent intramolecular nucleophilic substitution in refluxing EtOH to give thioflavones (Scheme
2(d)). Similarly, the reaction of 2’-methoxyalkynones with 2 equiv. of sodium sulfide in DMF afforded the thioflavones via 1,4-addition and subsequent substitution (Scheme 2(e)).18 The reaction of 2-(methylthio)benzoyl chloride and ary- lacetylenes in the presence of 2.5 equiv. of AlCl3 afforded the α,β-unsaturated β-chlorovinyl ketones in situ, which underwent acylation to give thioflavones (Scheme 2(f)).19 A palladium-catalyzed carbonylative four-component reac- tion was effective in the synthesis of thioflavones. Thus, the treatment of 2-iodofluorobenzene and arylacetylenes using sodium sulfide in the presence of 4 mol% Pd(OAc)2, 8 mol% t-Bu3P·HBF4, and 3 equiv. of Et3N under five bar of CO afforded the thioflavones (Scheme 2(g)).20
Although several types of reactions for the synthesis of thioflavones have been reported, some suffer from a lack of regioselectivity during cyclization, multiple steps, low yields, and harsh conditions. Previously, the condensation of S-(p-methoxybenzylthio) β-ketosulfoxides, derived from the protection of methyl 2-mercaptobenzoate and acyl substitution by sodium methylsulfinylmethide, with ary- laldehydes furnished the enones, which were then depro- tected and eliminated thermally to give the thioflavones, but it required five steps.21 This article describes the new synthesis of thioflavones by the regioselective cyclization of 1-(2-benzylthio)phenyl-3-phenylprop-2-yn-1-ones using hydrobromic acid.
N-Methoxy-N-methyl 2-mercaptobenzamide (2) was prepared using a previously reported method.22 Briefly, 3 equiv. of iso-PrMgCl were added to a mixture solution of methyl 2-mercaptobenzoate (1) and N,O-dimethylhydroxyl- amine hydrochloride in THF and 2 was obtained in 84%
yield after the usual acidic work-up (Scheme 3).TheS-ben- zylation of 2 was carried out by the treatment of 2 with sodium hydride in THF for 1 h at room temperature, fol- lowed by the addition of benzyl chloride and stirring for 1.5 h between 0oC and room temperature. After the usual aqueous work-up and purification by silica gel column Scheme 2. Several methods for the synthesis of thioflavones.
Scheme 3. Reagents and conditions: (a) CH3(CH3O)NH2Cl, 3 equiv. iso-PrMgCl, THF, -10-0 oC, 0.5 h; 1N HCl; (b) NaH, THF, rt, 1 h;
PhCH2Cl, 0 oC-rt, 1.5 h; (c) THF, 0 oC-rt, 0.5 h; 1 N HCl; (d) 2 equiv. 48 wt.% HBr, HOAc, rt or 60 oC, 1-3 h.
New Synthesis of Thioflavones by the Regioselective Cyclization of 1-(2-Benzylthio)phenyl-3-phenylprop-2-yn-1-ones Using Hydrobromic Acid 241
2020, Vol. 64, No. 4
chromatography, N-methoxy-N-methyl (2-benzylthio)ben- zamide (3) was obtained in 91% yield.
The synthesis of 1-(2-benzylthio)phenyl-3-phenylprop- 2-yn-1-ones (4a-l) was accomplished by the acyl substitution of 3 with arylethynyllithium compounds, which were gen- erated from arylacetylenes and CH3Li at 0oC, in THF for 0.5 h between 0oC and room temperature. After quenching the mixture with a 1 N HCl solution and the usual work-up, the residue was purified by silica gel column chromatog- raphy to give 4a-l in 84-91% yields.
The optimal conditions for the debenzylation and cycliza- tion of 1-(2-benzylthio)phenyl-3-phenylprop-2-yn-1- one (4b) were determined by screening different acids and sol- vents (Table 1). The reaction of 4b with 2 equiv. of 48 wt.%
HBr in HOAc at room temperature proceeded well to give thioflavone (5b) in 85% yield (entry 1). When CH3CN, DME, ClCH2CH2Cl were used as solvents, 5b was obtained in 91, 68, and 51% yield, respectively, after 6, 24, and 24 h, respectively, at room temperature (entries 2-4). On the other hand, the cyclization of 4b using 57 wt.% HI and 37 wt.% HCl in HOAc afforded 5b in 87 and 85% yields, respectively, after 1 and 24 h, respectively, at room tem- perature (entries 5, 6).
Thus, the regioselective 6-endo cyclization of 4a-l was carried out using 2 equiv. of 48 wt.% HBr in HOAc and the competitive 5-exo cyclization products were not observed in isolable amounts. The cyclization of 4a-l appeared to proceed through the debenzylation by HBr with the lib- eration of benzyl bromide to produce the corresponding 1- (2-mercapto)phenyl-3-phenylprop-2-yn-1-one intermedi- ates, which underwent rapid 1,4-addition to give thiofla- vones (5a-l). In general, the 6-endo cyclization of 4 using 2 equiv. of 48 wt.% HBr was completed within 3 h at room temperature. In contrast, the cyclization of alkynones with o-substituted aryl groups in the 3-position in 4 was com- pleted in 1-3 h at 60oC, reflecting the steric effect. The characteristic chemical shifts of the vinyl protons in 5 were
generally observed in the range of δ 7.18-7.25 ppm. On the other hand, the chemical shifts of the vinyl protons of 2’-substituted thioflavones (5c, 5f, 5h, 5k) were observed in the range of δ 6.19-7.17 ppm by the effect of dia- magnetic shielding due to the rotation of C1’-C2 single bonds.
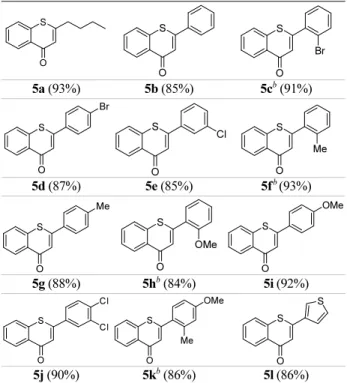
As listed in Table 2, various thioflavones were synthe- sized from 1-(2-benzylthio)phenyl-3-phenylprop-2-yn-1- ones using 48 wt.% HBr in HOAc in high overall yields (57-65%). The regioselective 6-endo cyclization of 4 worked well regardless of the types and positions of the electron- withdrawing (5c-e, 5j) and electron-donating (5f-i, 5k) substituents on the 2-substituted phenyl rings to give the corresponding thioflavones under the present conditions.
Furthermore, the reaction of 4 containing n-butyl and 3- thienyl groups proceeded equally well to give 2-(n-butyl)- 4H-benzothiopyran-4-one (5a) and 2-(3-thienyl)-4H-ben- zothiopyran-4-one (5l) in 93% and 86% yields, respec- tively.
In conclusion, the present method provides a highly regioselective synthesis of thioflavones via 6-endo cyclization of 1-(2-benzylthio)phenyl-3-phenylprop-2-yn-1-ones, derived from methyl 2-mercaptobenzoate, using 2 equiv. of hyd- robromic acid in HOAc in high yields.
Table 1. Effect of acids and solvents for the cyclization of 1-(2- benzylthio)phenyl-3-phenylprop-2-yn-1-one (4b)a
Entry Acidsb Solvents Time, h Yields of 5b, %c
1 HBr HOAc 1.5 85
2 HBr CH3CN 6 91
3 HBr DME 24 68 (25)
4 HBr ClCH2CH2Cl 24 51 (38)
5 HI HOAc 1 87
6 HCl HOAc 24 85
a The reaction was carried at room temperature. b 2 equiv. was used. c The numbers in parentheses indicate the recovery yields of 4b.
Table 2. Synthesis of thioflavones from 4 using hydrobromic acida
5a (93%) 5b (85%) 5cb (91%)
5d (87%) 5e (85%) 5fb (93%)
5g (88%) 5hb (84%) 5i (92%)
5j (90%) 5kb (86%) 5l (86%)
aThe conversion of 4 to 5 was carried out using 2 equiv. of 48 wt.% HBr in HOAc at room temperature for 1-3 h. bThe reaction was completed for 1-3 h at 60oC.
S
O
S O
S O
Br
S
O
Br S O
Cl S
O Me
S O
Me
S O
OMe
S O
OMe
S O
Cl
Cl S
O Me
OMe
S
O S
Journal of the Korean Chemical Society
242 Jae In Lee
EXPERIMENTAL
Preparation of 1-(2-benzylthio)phenyl-3-phenylprop- 2-yn-1-one (4b)
To a solution of N-methoxy-N-methyl (2-benzylthio) benzamide (3, 862 mg, 3.0 mmol) in THF (6 mL) was added lithium phenylacetylide, which had been generated from phenylacetylene (368 mg, 3.6 mmol) and CH3Li (1.5 M in Et2O, 2.4 mL, 3.6 mmol) in THF at 0 oC. The reaction mixture was stirred for 0.5 h between 0 oC and room temperature and quenched with a 1 N HCl solution (3 mL). After evaporating the solvents, the mixture was poured into a 0.1 N HCl solution (30 mL) and extracted with dichloromethane (3 × 20 mL). The organic layer was dried over anhydrous MgSO4 and filtered. The concen- trated residue was purified by silica gel column chroma- tography using 30% EtOAc/n-hexane as an eluting solvent to give 4b (887 mg, 90%). mp 120-121oC; 1H NMR (300 MHz, CDCl3) δ 8.42 (d, J = 7.8 Hz, 1H), 7.64-7.70 (m, 2H), 7.38-7.52 (m, 7H), 7.23-7.38 (m, 4H), 4.19 (s, 2H);
13C NMR (75 MHz, CDCl3) δ 177.7, 143.7, 135.9, 134.6, 133.4, 133.3, 133.0, 130.7, 129.2, 128.7 (overlapped), 127.4, 125.3, 123.7, 120.3, 92.9, 87.2, 37.1; FT-IR (KBr) 2200 (C≡C), 1623 (C=O) cm-1.
Preparation of thioflavone (5b)
Hydrobromic acid (48 wt.% in H2O, 453 μL, 4.0 mmol) was added to a solution of 4b (575 mg, 2.0 mmol) in HOAc (10 mL) and stirred for 1.5 h at room temperature. After evaporating of the HOAc, the reaction mixture was poured into a saturated NaHCO3 solution (30 mL) and extracted with dichloromethane (3 × 20 mL). The organic layer was dried over anhydrous MgSO4, filtered, and concentrated in vacuo. Benzyl bromide was evaporated further under a high vacuum, and the residue was recrystallized twice in 15% EtOAc/n-hexane to give 5b (405 mg, 85%). mp 125- 126 oC; 1H NMR (300 MHz, CDCl3) δ 8.55 (d, J = 7.9 Hz, 1H), 7.62-7.72 (m, 4H), 7.55-7.61 (m, 1H), 7.48-7.54 (m, 3H), 7.25 (s, 1H); 13C NMR (75 MHz, CDCl3) δ 180.9, 153.1, 137.7, 136.6, 131.6, 130.9, 130.8, 129.3, 128.6, 127.8, 127.0, 126.5, 123.5; FT-IR (KBr) 1615 (C=O) cm-1; Ms m/z (%) 238 (M+, 100). The physical andspectral data for the thio- flavones associated with this article can be found in the Supporting Information.
Supporting Information. Additional supporting infor- mation may be found online in the Supporting Informa- tion section at the end of the article.
REFERENCES
1. Nakazumi, H.; Ueyama, T.; Kitao, T. J. Heterocyclic Chem.
1984, 21, 193.
2. Zhang, D.; Ji, X.; Gao, R.; Wang, H.; Meng, S.; Zhong, Z.; Li, Y.; Jiang, J.; Li, Z. Acta Phar. Sin. B 2012, 2, 575.
3. Choi, E. J.; Lee, J. I.; Kim, G.-H. Int. J. Mol. Med. 2012, 29, 252.
4. Wang, H.-K.; Bastow, K. F.; Cosentino, L. M.; Lee, K. H. J.
Med. Chem. 1996, 39, 1975.
5. Jang, E. J.; Seok, Y. M.; Lee, J. I.; Cho, H. M.; Sohn, U.
D.; Kim, I. K. Naunyn-Schmiedeberg’s Arch. Pharmacol.
2013, 386, 339.
6. For reviews, see: (a) Dong, J.; Zhang, Q.; Meng, Q.; Wang, Z.; Li, S.; Cui, J. Mini-Reviews Med. Chem. 2018, 18, 1714. (b) Sosnovskikh, V. Y. Russ. Chem. Rev. 2018, 87, 49.
7. (a) Nakazumi, H.; Watanabe, S.; Kitaguchi, T.; Kitao, T.
Bull. Chem. Soc. Jpn. 1990, 63, 847. (b) Sabatini, S.;
Gosetto, F.; Manfroni, G.; Tabarrini, O.; Kaatz, G. W.;
Patel, D.; Cecchetti, V. J. Med. Chem. 2011, 54, 5722.
8. (a) French, K. L.; Angel, A. J.; Williams, A. R.; Hurst, D.
R.; Beam, C. F. J. Heterocyclic Chem. 1998, 35, 45. (b) Metz, C. R.; Knight, J. D.; Pastine, S. J.; Pennington, W.
T.; Beam, C. F. J. Chem. Crystallogr. 2010, 40, 536.
9. Vijay, T. A. J.; Nandeesh, K. N.; Raghavendra, G. M.;
Rangappa, K. S.; Mantelingu, K. Tetrahedron Lett. 2013, 54, 6533.
10. Lee, J. I. Bull. Kor. Chem. Soc. 2009, 30, 710.
11. Vargas, E.; Echeverri, F.; Velez, I. D.; Robledo, S. M.;
Quinones, W. Molecules 2017, 22, 2041.
12. (a) Kumar, P.; Rao, A. T.; Pandey, B. J. Chem. Soc. Chem.
Commun. 1992, 1580. (b) Kumar, P.; Bodas, M. S. Tetra- hedron 2001, 57, 9755.
13. Kobayashi, K.; Kobayashi, A.; Ezaki, K. Heterocycles 2012, 85, 1997.
14. Sangeetha, S.; Sekar, G. Org. Lett. 2019, 21, 75.
15. Wang, D.; Sun, P.; Jia, P.; Peng, J.; Yue, Y.; Chen, C. Syn- thesis 2017, 49, 4309.
16. (a) Fuchs, F. C.; Eller, G. A.; Holzer, W. Molecules 2009, 14, 3814. (b) Lee, J. I. Bull. Kor. Chem. Soc. 2012, 33, 1375.
(c) Lee, J. I.; Choi, J. S. J. Kor. Chem. Soc. 2015, 59, 253.
17. (a) Willy, B.; Muller, T. J. J. Synlett 2009, 2009, 1255. (b) Willy, B.; Frank, W.; Muller, T. J. J. Org. Biomol. Chem.
2010, 8, 90.
18. Yang, X.; Li, S.; Liu, H.; Jiang, Y.; Fu, H. RSC Adv. 2012, 2, 6549.
19. Kim, H. Y.; Song, E.; Oh, K. Org. Lett. 2017, 19, 312.
20. Shen, C.; Spannenberg, A.; Wu, X. F. Angew. Chem. Int.
Ed. 2016, 55, 5067.
21. (a) Taylor, A. W.; Dean, D. K. Tetrahedron Lett. 1988, 29, 1845. (b) Kataoka, T.; Watanabe, S.; Mori, E.; Kadomoto, R.; Tanimura, S.; Kohno, M. Bioorg. Med. Chem. 2004, 12, 2397.
22. Lee, J. I. J. Kor. Chem. Soc. 2019, 63, 398.