Vol. 15, No. 2, November, 2007
□ 종 설 □1)
서 론
포도당은 포유동물 대사에 필수적인 에너지원이 며 특히 뇌신경세포에 중요한 에너지원이다. 당뇨병 이 알려진 이래로 포도당 대사장애는 임상 의사들 의 주요 관심사였으며 지속되는 연구로 포도당과 인슐린이 주요 역할을 하는 호르몬 간의 관계가 알
책임저자 : 김성구, 한림의대 강남성심병원 소아청소년과학교실 Tel : 02)829-5144, Fax : 02)845-5144
E-Mail : [email protected]
려지고 포도당 수송에 중요한 역할을 하는 2가지 종류의 수송자가 밝혀졌는데 하나는 능동수송을 하 는 Na+-glucose cotransporter이며 다른 하나는 조직장벽을 확산에 의해 통과하도록 하는 촉진성 포도당 수송자(facilitative glucose transporter)가
있다1, 2). 이 두 수송자에 대한 이해가 이루어지면서
기존에 알려진 Fanconi-Bickel syndrome과 새롭 게 알려지기 시작한 Glucose-galactose malabsorp- tion syndrome, 포도당 수송자-1 결핍증에 대한 분자 유전학적 이해가 이루어지게 되었다.
포도당 수송자-1 결핍 증후군
한림대학교 의과대학 소아과학교실
김 성 구
= Abstr act =
Glucose Transporter Type 1 Deficiency Syndrome
Sung Koo Kim, M.D.
Department of Pediatrics, College of Medicine, Hallym University, Seoul, Korea
D-glucose is an essential fuel for metabolism in mammalian cells and the predominant fuel source for the brain. Transport of glucose across tissue barriers is mediated by ste- reospecific transporter proteins. Glut-1 is a major glucose transporter expressed on vas- cular endothelial cells comprising the blood brain barrier and is responsible for glucose entry into the brain. Impaired glucose transport across the blood brain barrier results in Glut-1 deficiency syndrome(DS). It is caused by haploinsufficiency of the blood brain barrier hexose carrier. Heterozygous mutations or hemizygosity of the GLUT-1 gene cause Glut-1 DS. It is characterized by infantile seizures refractory to anticonvulsants, develop- mental delay, acquired microcephaly, spasticity, ataxia, opsoclonus and other paroxysmal neurological phenomena, often occurring prior to meals. The diagnosis of Glut-1 DS is established in neurologically impaired patients with reduced cerebrospinal glucose concent- ration(hypoglycorrhachia) and lactate concentration in the absence of hypoglycemia. De- creased 3-O-methyl-D-glucose uptake in erythrocytes also supports the diagnosis of Glut-1 DS. Several treatment strategies have been pursued, none optimal, as it relates to the developmental encephalopahty associated with this clinical syndrome. Ketogenic diet has been effective in controlling seizures but has had little measurable effects on the associated cognitive impairments and behavioral disturbance. Current treatment is inade- quate, and future studies should be directed at the mechanisms designed to upreglulate GLUT-1 expression, thereby increasing residual Glut-1 activity to 75 to 100%.
Key Words : Glut-1 deficiency syndrome, Epilepsy, Hypoglycorrhachia
1985년 Mueckler 등3)에 의해서 인간 HepG2 간 암세포에서 포도당 수송자-1 단백질이 추출되었으 며 이후 조직 분포, 세포내 위치, 수송역학(trans- port kinetics) 등에 따라서 12가계의 포도당 수송 자가 밝혀졌다4-7). 포도당 수송자-1은 뇌혈관 관문 을 구성하는 혈관내피세포와 성상세포 막에 주로 위치하여 뇌로의 포도당 이동의 관문 역할을 하고 있다. 포도당 수송자-1의 유전자 변이에 의한 포도 당 수송자-1 결핍 증후군은 1991년 De Vivo8, 9)에 의해서 영아기에 시작되는 간질성 뇌증후군과 동반 되는 진행성의 두위 감소에 의한 후천성 소두증, 운 동실조와 강직을 보인 두 명의 환자를 새로운 질환 으로 기술함으로써 알려지기 시작하였다. 전 세계적 으로 100여례가 보고 되었으며 최근 일본과 홍콩을 포함한 아시아에서도 발견되고 있다.
본 고에서는 난치성 간질의 한 원인이며 대뇌 포 도당 대사 장애질환으로 최근 알려지기 시작한 포 도당 수송자-1 결핍 증후군에서 지금까지 밝혀지고 연구된 내용들을 알아보고자 한다.
본 론
1. 포도당 수송과 대사
포도당 수송자-1 결핍 증후군 환자의 연구를 통 해서 알려진 바로는 뇌 대사를 위해서 포도당이 뇌 혈관 내피 세포와 성상세포막으로 구성된 뇌혈관 관문을 통과하는 것이 가장 중요한 수송 과정(rate limiting step)이며10) 여기에 주로 존재하는 수동적 촉진 수송자인 포도당 수송자-1이 이를 주로 담당 하고 있음이 증명되었다. 포도당은 뇌로 들어간 후 에 비가역적으로 인산화되어 glucose-6-phosphate 로 전환된 후 pentose phosphate shunt나 Emb- den-Meyerhoff pathway에서 대사 되거나 글리코 겐으로 전환된다. Pentose phosphate shunt는 핵 산 합성에 중요하며 Embden-Meyerhoff pathway 는 포도당을 대사과정을 통해 pyruvate로 변환시킨 다. 글리코겐의 합성은 대사 스트레스 기간에 에너 지원으로 사용될 수 있도록 저장된다. 뇌로의 포도
당 수송 감소는 이와같은 세가지 경로를 제한해서 포도당 수송자-1결핍 증후군을 일으키게 된다.
이전에는 모든 경우에 있어서 뇌 대사에는 포도 당 만이 유일한 에너지원으로 생각되어 왔으나 1967년 만성적인 케톤혈증이 유발된 상태에서는 케 톤체가 포도당을 부분적으로 대치 할 수 있다는 것 이 밝혀졌다11). 케톤체는 포도당 수송자를 거치지 않고 단카르복실 수송체(monocarboxylic transpor- ter-MCT)를 통하여 뇌로 들어가게 되며 미토콘드 리아 기질안에서 acetyl-CoA로 대사된다12).
케톤체는 포도당에서 pyruvate의 합성이 제한되 는 포도당 수송자-1 결핍증, pyruvate dehydro- genase deficiency와 그 밖의 뇌 포도당 대사 장애 질환에서 acetyl-CoA의 공급원이 된다13).
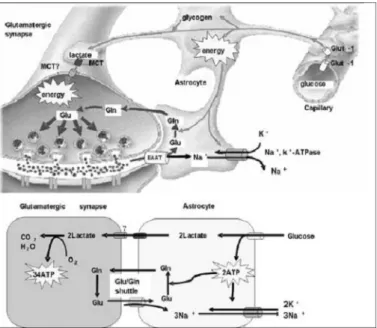
포도당 수송자-1 결핍 증후군의 병태생리를 이 해하는데 많은 기여를 한 Magistretti의 뇌 포도당 대사에 관한 새로운 이론에 따르면 포도당 수송자- 1에 의해서 성상세포에 흡수된 포도당이 lactate로 대사되어 신경세포에 공급되어 에너지원으로 이용 된다는 것이며14) 또한 신경세포에서 발견되는 포도 당 수송자-3이 신경세포의 포도당 흡수에 일정 부 분 역할을 하는 것으로 생각되고 있다15)(Fig. 1).
포도당 수송자-1 결핍 증후군과 같이 포도당의 뇌 혈관관문과 성상세포막 통과가 제한되는 경우에는 성상세포에서 글리코겐의 저장과 lactate의 합성이 어려워지게 되며 이는 신경세포의 에너지 이용에 제한을 가져오게 된다. 이와 같은 이해는 포도당 수 송자-1 결핍증 환자에서 관찰되는 lactate 감소에 합당한 소견이다.
2. 포도당 수송자의 분자유전학적 구조와 생리 8개의 포도당 수송자 가계는 각각 다른 조직과 세포 내에 분포하며 특징적 수송 역학(transport kinetics)을 보인다. 포도당 수송자-1 유전자는 염 색체 1번의 단완에 위치하며16) 35 kb 크기로 10개 의 엑손과 9개의 인트론으로 구성되어 있고 492개 의 아미노산으로 이루어진 단백질을 지정한다17). 포 도당 수송자-1은 HepG2/erythrocyte/brain trans- porter로 불리며 포도당 수송자 가계 중 처음으로
밝혀진 수송자이다. 대부분의 조직에 존재하지만 적 혈구, 뇌 미세혈관, 성상세포에서 주로 발현된다. 포 도당 수송자-1은 12개의 막 내외 도메인(trans- membrane domains)로 구성되어 있으며 45 kDa와 55 kDa 분자량의 2가지 종류가 있는데 같은 유전
자에 의해서 발현되며 단지 포도당화에만 차이가 있다18)(Fig. 2). 55 kDa은 주로 뇌 미세혈관 내피 세포와 적혈구 막에 주로 존재하며 45 kDa은 아교 세포, 신경세포, 맥락막총(choroid plexus)에 위치 하여 이들 조직에 포도당 흡수를 담당한다15).
포도당 수송자-1은 주로 포도당을 촉진 수송하 지만 그밖에도 물19), 갈락토오스1, 20), dehydroascor- bic acid(DHA)21), glycopeptide22) 등이 수송되며 DHA는 뇌혈관 관문을 통과한 후 ascorbic acid로 전환된다23).
3. 임상 양상
이완된 영아는 임신과 출산 시 특별한 문제가 없 으며 체중과 Apgar score가 정상이다. 일반적으로 환자는 간질성 뇌증후군과 동반되어 신경발달지연, 진행성 두위 감소에 의한 후천성 소두증, 운동실조 와 강직을 보이게 된다. 포도당 대사의 견지에서 살 펴보면 뇌의 주요 에너지원은 포도당이며 이는 포 Fig. 1. The concept of the astrocyte-neuron lactate shuttle.
The hypothesis is modified to accomodate the classification of brain energy failure syndrome. Central to the hypothesis is the shuttling of lactate from the astrocyte to the neuron(Magistretti et al., Science 283, 496[1999]. Copyright [1999] American Asso- ciation for the Advancement of Science.). Abbreviations : ATP, adenosine triphosphate; EAAT, excitatory amino acid trans- porter; Gln, glutamine; Glu, glutamate.
Fig. 2. Glucose transporter-1 consists of 12 trans- membrane domain
도당 수송자-1에 의해서 뇌로 들어갈 수 있다. 뇌 의 포도당 대사율은 태아기와 출생 직후에는 낮지 만 출생 후 지속적으로 증가해서 3세 전후에 최고 조에 달한 후 10세까지 일정하게 유지되고 이후 성 인기까지 서서히 감소하게 된다24). 따라서 포도당 수송자-1 결핍 증후군의 임상증상은 태아기와 출생 직후에는 보이지 않고 영아기와 초기 소아기에 주 로 증상을 보이게 된다.
1) 경 련
경련은 첫 번째 뇌기능 장애의 증상으로서 일반 적으로 1개월에서 4개월 사이에 일어나며 무호흡 발작, 안구경련(opsoclonus)과 유사한 비정상적인 안구운동 등의 국소발작 형태로 나타나며 뇌파소견 은 다 촛점성 극파를 나타낸다. 뇌가 성숙되어 가면 서 경련은 동기화되고 전신발작의 형태로 나타나게 되며 뇌파는 비전형적인 3-4 Hz 극서파를 보이게 된다. 경련 형태는 주로 전신성 강직, 간대, 근간대, 비전형 소발작, 탈력발작 등의 형태로 나타나게 된 다. 경련의 빈도는 환자에 따라 하루 수차례부터 수 개월에 한차례까지 다양하며 대부분의 환자에서 경 련을 보이지만 매우 드물게 경련이 없을 수도 있다.
2) 기타 돌발성 장애
간헐적인 운동실조, 의식혼미, 기면, 부전마비, 근 긴장 이상과 같은 비정상적인 운동과 자세, 체간마 비, 수면장애와 반복적인 두통이 있을 수 있다. 이 와 같은 증상이 간질성인지 비간질성인지는 확실치 않으며 심한 변동성을 띠고 금식과 피로 등에 의해 서 영향을 받는다. 모든 환자들은 다양한 정도의 언 어장애를 가지고 있으며 유창성 장애(dysfluency) 와 관련된 구음장애가 흔한다. 수용언어와 표현언어 모두 장애가 있으나 표현언어의 장애가 좀 더 심하 다. 다양한 정도의 인지기능 장애를 보여 가벼운 학 습장애부터 심각한 지능장애를 보이기도 한다. 사회 적응행동은 다른 장애에 비해서 예외적으로 좋아서 집단 활동이나 학교생활에 비교적 잘 적응한다.
3) 유전자-표현형의 관련성(Genotype-Phen otype Correlation)
유전자 변이에 따른 표현형의 분류는 뇌혈관 관 문의 포도당 수송자-1의 기능정도에 따라서 다음과
같은 가설이 제시되고 있다25).
(1) 최소 표현형(minimal phenotype)을 보이는 경우는 75-100%의 포도당 수송자-1의 기능이 남 아 있는 경우로 과오 돌연변이에 의한 기능감소 혹 은 포도당 수송 억제 효과가 있는 에탄올26, 27), 카페 인28), 약물29)투여에 의한 경우이다.
최소 표현형의 경우 임상증상을 보이는 경우는 드물지만 발열이나 다른 환경적 인자에 의해서 유 발되어 경미한 일과성 증상을 보일 수 있다.
(2) 경미한 표현형(mild phenotype)을 보이는 경 우는 50-75%의 포도당 수송자-1의 기능이 보존되 어 있는 경우로 이종 과오 돌연변이(heterozygous missense mutation)의 경우이다.
(3) 중증도 표현형(moderate or classic pheno- type)을 보이는 경우는 넌센스 돌연변이(nonsense mutation), 해독틀이동 돌연변이(frameshift muta- tion), 반접합성(hemizygosity), 절단부위 돌연변이 (splice-site mutation)등이며 포도당 수송자-1 단 백질이 50% 이상 감소되어 있는 경우이다.
(4) 중증 표현형(severe phenotype)은 포도당 수 송자-1의 기능이 25-50% 남아있는 경우로 복합이 종 돌연변이(compound heterozygosity mutation)
30)의 경우로 미세한 증상을 보이는 부모로부터 돌 연변이 유전자를 받고 대립 유전자(allele) 돌연변이 가 복합된 경우로 중증의 증상을 나타내게 된다.
(5) 태아사망(embryonic lethal)은 포도당 수송 자-1의 기능이 0-25% 정도 남아 있는 경우로 동형 접합 돌연변이에 의하며 포도당 수송자-1은 syncy- totrophoblast와 cytotrophoblast 세포막에 발현되 는 주요 수송자이므로28) 태아에 포도당 공급중단이 일어나게 된다31, 32).
4. 유 전
포도당 수송자-1 결핍 증후군은 상염색체 우성 으로 유전되나 환자의 부모는 드물게 포도당 수송 자-1 결핍 증후군에 이환되어 있으며 질환의 정도 는 경미하거나 무증상인 경우가 많다. 대부분의 환 자는 새로운 돌연변이에 의하며 가족력이 있는 경 우에는 세대가 내려갈수록 질환의 중증도가 증가한
다. 이환율은 질환의 기술 자체가 오래되지 않아 정 확히 알 수 없으나 전세계적인 발견 보고가 있다.
5. 검사 소견
임상적으로 의심되는 경우에 뇌척수액 검사는 필 수적인데 혈당은 정상인데 비해 척수액내 포도당 농도는 감소(hypoglycorrhachia)를 보이며 일반적 으로 40 mg/dL 이하로 대개 30 mg/dL 중반의 수 치를 보인다33). 뇌척수액의 포도당 농도는 4시간 금 식 후에 측정하고 혈당은 척수액 검사 직전에 측정 하게 되는데 혈당에 대한 뇌척수액의 당농도 비는 약 0.33±0.01(정상: 0.65±0.01)를 보인다. lactate 농도는 정상 혹은 경미하게 감소한 검사치를 보여 서 대개 1.4 mM 이하의 소견을 보인다.
뇌자기공명영상(MRI)과 전산화단층촬영술(CT) 은 일반적으로 정상소견을 보이지만 일부환자에서 경미한 뇌 위축소견을 보인다. 양전자방사단층촬영 (PET)은 전반적인 포도당 섭취 감소를 보이며 특 히 내측 측두엽과 시상부위에서 좀 더 심한 감소를 보인다. 반면에 바닥핵 부위는 상대적으로 포도당 흡수가 유지되는 소견을 보이는데 이와 같은 불일 치는 치료 혹은 질환의 심한 정도와 상관없이 성인 기까지 유지된다34)(Fig. 3). 이 검사는 진단에 보조 적 수단으로 사용될 수 있으나 민감도와 특이도는 아직 명확하지 않다.
적혈구 포도당 흡수도 검사는 유용한 선별검사방 법이며 임상적으로 의심되는 모든 환자에서 실시해 야 한다. 적혈구의 포도당 수송자-1의 단백질은 면
Fig. 3. PET scan, patients were fasted for at least 6h prior to the injection of the radiopharmaceutical. Patient blood glucose levels were measured at the time of the study and found to be normal. Intravenous access was obtained at least 15 min prior to the radiopharmaceutical administration. The patients were then injected with 0.14 mCi/Kg of 18F-2-deoxyglucose and scanned 30 min following injection. Studies were acquired on a Siemens ECAT EXACT HR(+), (Knoxville, TN) with FWH=4 mm. Each study was acquired using a multiframe technique and auto attenation correction in a dynamic scan mode (4 frames at 480 sec per frame) that used filter back projection. Reconstruc- tion with auto attenuation correction used a Hann filter(cutoff 0.40 cycles/
pixel). Post reconstruction transverse, oblique transverse, coronal, and sagittal plane images with a slice thickness of 0.50 cm were then produced and dis- played using an inverted gray scale and rainbow(16 step) maps. PET scan studies in Glut1 deficiency syndrome patients have shown a global reduction of glucose uptake in the brain, with more severe hypometabolism in the medial temporal lobes and the thalami(De Vivo DC et al., Glucose metabolism in the brain, San Diego: Academic Press, 2002:274).
역화학적으로 뇌혈관의 것과 동일하다. 신선하게 분 리되어 세척된 적혈구에서 3-O-methyl-D-glucose 의 흡수도는 정상인과 비교 시 환자에서 약 50%정 도 감소되는 것을 볼 수 있다. 위 검사는 포도당 수 송자-1 결핍 증후군의 진단에 매우 예민하고 특이 적이지만 위음성이 있을 수 있으며 당뇨병을 포함 한 만성적인 고혈당 상태에 있는 환자의 경우에는 세포막에 위치한 포도당 수송자-1 단백질의 하향조 절(downregulation)에 의해 위양성이 나올 수도 있 다35).
림프구나 피부 섬유세포를 배양해서 FISH(fluo- rescence in situ hybridization)를 이용하여 큰 결 실을 가진 경우를 진단 할 수 있다36). 현재 3명의 포도당 수송자-1 결핍환자가 이 검사로 반접합체 (hemizygous)로 진단되었다(Fig. 4).
FISH검사가 음성인 경우에 포도당 수송자-1에 대한 유전자 분석을 시행한다. Genomic DNA는 환자의 혈액이나 배양된 피부 섬유세포로부터 추출 하여 이용한다. 현재까지 알려진 돌연변이는 30례 의 이접합성 돌연변이로 과오 돌연변이 18례, 넌센
스 돌연변이 3례, 삽입 3례, 극소결실 3례, 절단부위 돌연변이 3례를 포함하고 있다25)(Fig. 5).
6. 감별 진단
간헐적인 저혈당증에 의해서 신경저혈당(neuro- glycopenia)를 보이는 가족성 고인슐린 혈증(famil- ial hyperinsulinism), 신생아 경련과 후천성 소두 증의 모든원인 특히 렛트 증후군(Rett syndrome), 엔젤만 증후군(Angelman syndrome), 영아형 neu- ronal ceroid lipofuscinosis, opsoclonus-myoclonus syndrome과 감별해야 하며 발달지연이 있는 잠재 성 간질성 뇌증후군, 상염색체 우성유전하는 가족성 간질, 돌발성 신경장애로 교대성 반신부전마비, 실 조, 경련, 근긴장이상증을 포함하는 운동장애와 인 지기능 장애를 보이면서 당분섭취로 호전되는 경우 감별진단에 포함된다.
7. 치 료
포도당 수송자-1 결핍 증후군 환자에서 뇌혈관 관문을 통한 포도당 수송 감소가 확인되기 전인
Fig. 4. Fluorescence in Situ hybridization(FISH). Metaphase spreads of cultured skin fibroblasts from patients were probed with labeled p1 DNA specific for GLUT1. Hemizygosity has been found in 0-15% of Glut-1 DS patients. (A) A FISH-positive Glut-1 DS patients who is hemizy- gous for GLUT1 is indicated by arrows with normal and deletion. (B) A FISH-negative Glut-1 DS patients who carries a heterogyous deletion(1186delG) is indicated by arrows with normal(De Vivo DC et al., Glucose metabolism in the brain, San Diego: Academic Press, 2002:275).
1991년에 이미 처음으로 환자에서 케톤식이 요법이 도입되었다37). 케톤체는 조직막을 포도당 수송자-1 대신 MCT을 통해서 이동함으로12) 포도당 수송자- 1이 결핍된 경우에 대체 에너지원으로서 뇌로 흡수 되고 뇌세포에서 직접 이용할 수 있게 된다. 케톤식 이는 경련의 조절에는 매우 효과적이지만 뇌에서 케톤체 이용 부위에 대해서는 확실히 밝혀져 있지 않으며 인지와 사회적응을 포함하는 신경행동장애 에 효과가 있는지 여부는 확실치 않다38, 39). 또한 대 부분의 환자에서 영아기에 두위감소로 삼 백분위수 이하의 소두증을 보이게 되는데 어떤 뇌 구조물이 주요 위험 부위인지 확실하지 않지만 성상세포의 증식에 의한 수초화(myelination), 수상돌기의 분지 형성(dendritic arborization)과 시냅스형성(synap- togenesis)이 출생 후에도 진행됨으로 포도당 수송
장애에 의한 에너지 결핍에 의해서 이와 같은 과정 이 손상을 받을 수 있으며 만성적인 케톤혈증이 이 중 일부 손상만을 막을 수 있는 것으로 생각되고 있다9).
Alpha-lipoic acid(thioctic acid)는 배양된 근육 세포에서 세포기질내의 포도당 수송자-4를 세포막 으로 이동시켜 포도당 수송을 촉진시키는 것으로 알려지고 있으며40) 환자의 섬유아세포에서는 포도 당 수송자-1의 기능을 증가시키는 것으로 보고되었 다41). 따라서 thioctic acid의 신경세포 포도당 수송 자-1에 대한 확실한 연구 데이터는 없지만 위와 같 은 결과에 근거해서 환자에게 thioctic acid를 추천 해 왔으나42) 효과는 확실치 않으며 적절한 치료 농 도에 도달하게 위해서는 경구 투여로 충분치 않고 고용량을 투여해야 하며 이 경우 위장관계 부작용 이 있을 수 있다41).
조직 장벽을 통한 포도당의 수송은 여러 가지 다 른 인자에 의해서도 영향을 받을 수 있는데 만성적 인 고혈당은 포도당 수송자를 하향 조절하는 원인 이 되는 반면에 저혈당이나 급성 저산소증은 포도 Fig. 5. Summary of the mutations identified in
the GLUT-1 gene causing Glut-1 deficiency synd- rome. Filled boxes represent the 10 exons and the lines represent the introns. Missense mutations are listed on the left side of the gene structure;
the other mutations including nonsense, insertion, deletion, and splice-site mutations are listed on the right side. The three patients with hemizy- gosity are not represented. The superscript num- bers represent the number of patients sharing the same mutation. Asterisk denotes mutations identi- fied by others(Dong Wang et al., Glut-1 deficiency sydrome: clinical, genetic, and therapeutic aspects.
Ann Neurol 2005;57:111-8).
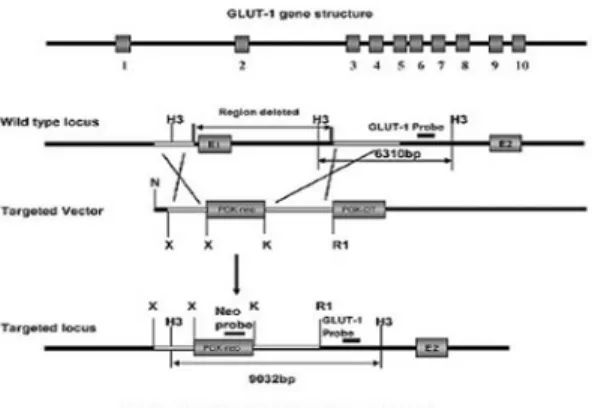
Fig. 6. Generation of Glut-1-deficiency mice by homologous recombination. Gene structure of the mouse GLUT-1 gene, restriction map of the tar- geted region, targeting vector used for the homo- logous recombination and the targeted locus. Gene targeting resulted from deletion of promoter region and exon 1 and insertion of the PGK-Neo fusion cassette. Gray boxes represent the GLUT-1 exons.
Restriction enzymes: H3, hindIII; K, kpnI; N, notI;
R1, ecoRI; X, XhoI. The GLUT-1 probe and Neo probe are indicated(Wang D, et al., A mouse model for Glut-1 haploinsufficiency. Hum Mol Genet 2006;
15:1169-79).
당 수송자-1을 상향 조절시킨다. 그 밖에도 여러 가지 인자들이 포도당 수송에 영향을 미치는데 영 아기에 시작되는 경련에서 가장 많이 쓰이는 항경 련제 중의 하나인 phenobarbital이 포도당 수송자- 1 결핍증에 사용되었을 때 대부분의 환자에서 경련 이 조절되지 않고 임상상태가 악화되는 것이 보고 되고 있다. 생체외(In vitro) 실험에서 포도당 수송 자-1 결핍 증후군 환자 적혈구에서 포도당 수송은 barbiturate에 의해서 악화되는 것으로 밝혀졌고43,
44) methylxanthine도 포도당 수송을 억제하는 것 으로 나타났다28). 따라서 환자에서 barbiturate, methylxanthine을 피해야 하며 커피와 카페인이 들어있는 음료도 마시지 않을 것을 추천한다45).
포도당 수송자-1 결핍증이 의심되는 모든 환자 는 케톤식이 요법이 시도되지만 청소년기와 어른 환자에서는 순응도가 떨어져 경련이 재발하고 증상 이 나타나더라도 일반식이로 되돌아가는 경향이 있 다. 따라서 다른 식이요법에 대한 연구가 필요한데 비교적 경미한 임상증상을 가진 환자를 대상으로 높은 혈중 포도당 농도의 유지를 통한 포도당 수송 증가 효과를 위해 요리되지 않은 cornstarch가 강 화된 고혈당식이와 diazoxide 같은 약물의 병합요 법이 시도되고 있다. diazoxide는 sulfonyl urea re- ceptor-potassium channel complex를 개방해서 췌장 도세포(islet cell)을 과분극시켜 인슐린 분비 를 억제하여 혈당을 증가시키는데 식간 혈당 농도 를 증가시키는 효과가 있다. 글루카곤 또한 포도당 신생합성과 글리코겐 분해, 근육과 간에서 포도당 흡수 억제를 통해서 혈당을 증가시키는데 급성기 돌발성 증상의 치료에 효과적이다25).
8. 포도당 수송자 결핍 질환의 마우스 모델 여러 종류의 마우스 모델이 포도당 수송자 돌연 변이에 의한 병태생리를 밝히기 위해서 만들어졌으 며 동형 접합자 포도당수송자-2 유전자 파괴에 의 한 모델에서는 고혈당과 저인슐린혈증이 유발되었 다46). 포도당 수송자-2는 췌장기능의 유지와 정상 혈당의 항상성 유지를 위해서 필수적이며 인간의 Fanconi-Bickel 증후군의 모델이다. 포도당 수송자
-4 녹아웃(knock out) 마우스모델은 인슐린 비의존 성 당뇨병과 유사하여 근육의 포도당 흡수가 감소 되며 고혈압을 유발하고, 간과 심장에 질환을 일으 킨다47).
포도당 수송자-1 결핍 증후군 마우스 모델의 개 발은 최근까지 성공적이지 못했으나 최근 컬럼비아 대학 소아 신경 Colleen Giblin lab에서 targeted disruption으로 GLUT+/- 녹아웃 마우스가 확립되 었다(Fig. 6).
이 마우스 모델은 인간 포도당 수송자-1 결핍 증 후군과 매우 유사하게 경련, 뇌척수액의 포도당 감 소, 운동장애, 조화불능, 학습장애, 소두증, 양전자방 사단층촬영(PET)에서 감소된 포도당 흡수, 뇌조직 에서 포도당 수송자-1 단백질의 감소를 보여 향후 에 포도당 수송자 결핍 증후군의 신경화학, 병리, 행동 장애에 대한 연구를 할 수 있을 것으로 기대 되고 있다48).
9. 향후 연구 방향
De Vivo에 의해서 1991년 처음으로 포도당 수 송자-1 결핍 증후군이 기술된 이래로 1998년 포도 당 수송자-1 유전자 돌연변이가 질환의 원인으로 확인되었으며 2006년 인간의 질환과 유사한 임상증 상을 나타내는 녹아웃 마우스가 확립되었다. 이 모 델을 통해서 질환의 병태생리에 대한 연구와 후천 적 소두증에 대한 세포학적 구조적 연구를 할 수 있을 것으로 기대하고 있다. 상염색체 열성 유전에 의한 포도당 수송자-1 유전자 돌연변이로 질환이 발현됨으로 약물치료의 목표는 존재하는 정상 대립 유전자의 상향조절에 맞추어지고 있다. 이와 같은 치료법 개발에 성공한다면 유사한 다른 우성유전 질환의 치료제 연구에 모델이 될 수 있을 것이다.
또 다른 연구 방향으로는 진행하는 소두증의 병태 생리의 한 과정이 신경세포와 글리아세포의 자멸사 로 생각되고 있음으로 이의 차단에 의한 치료가 주 요 관심사의 하나이다. 현재까지 약제로는 환자의 피부 섬유아세포에서 thioctic acid가 포도당 수송 자-1의 기능을 증가시키는 것으로 알려지고 있으며 녹아웃 마우스를 이용한 실험에서 뇌혈관 관문을
구성하는 세포중 하나인 성상세포를 분리 배양하여 이들 세포에 다양한 약제를 투여하여 포도당 수송 자-1의 기능을 증가시키는 연구가 시도되고 있다.
결 론
포도당 수송자-1 결핍 증후군은 드물고 새롭게 알려지기 시작한 질환이지만 동남아시아를 포함한 전 세계적인 환자의 발견 보고의 증가와 표현형의 다양성을 고려하여 경미한 증상을 보이는 경우까지 포함 하면 좀더 많은 환자군이 있을 것으로 추정되 고 있다. 난치성 간질과 소두증을 보이는 발달장애 의 한 원인으로 본 질환에 대한 관심이 필요하며 적절한 진단과 케톤식이 요법으로 경련의 조절과 인지기능, 운동기능 장애에 대해 일부 도움을 받을 수 있다. 또한 최근에 확립된 녹아웃 마우스를 이용 한 연구로 질환에 대한 정확한 병태생리를 밝히고 치료제 개발에 대한 연구가 활성화 될 것으로 기대 되고 있다.
References
1) Bell IG, Burant CF, Takedda J, Gold GW.
Structure and function of mammalian facilita- tive sugar transporters. J Biol Chem 1993;268:
19161-4.
2) Longo N, Elsas LJ. Human glucose transpor- ters. Adv Pediatr 1998;45:293-313.
3) Mueckler M, Caruso C, Baldwin SA. Sequence and structure of a human glucose transporter.
Science 1985;229:941-5.
4) Bell GI, Kayano T, Buse JB. Molecular biology of mammalian glucose transporters. Diabetes Care 1990;13:198-208.
5) Baldwin SA. Mammalian passive glucose trans- porters: members of an ubiquitous family of active and passive transport proteins. Biochim Biophys Acta 1993;1154:17-49.
6) Doege H, Schurmann A, Bahrenberg G.
GLUT8, a novel member of the sugar transport facilitator family with glucose transport acti- vity. Biol Chem 2000;275:16275-80.
7) Doege H, Bocianski A, Joost HG, Schurmann A. Activity and genomic organization of
human glucose transporter 9(GLUT9), a novel member of the family of sugar-transport facili- tators predominantly expressed in brain and leukocytes. Biochem J 2000;350:771-6.
8) De Vivo DC, Trifileti RR, Jacobson RI. Defec- tive glucose transport across the blood-brain barrier as a cause of persistent hypoglycorr- hachia, seizures, and developmental delay. N Engl J Med 1991;325:703-9.
9) De Vivo DC, Garcia-Alvarez M, Ronen GM.
Glucose transport protein deficiency: an emer- ging syndrome with therapeutic implications.
Int Pediatr 1995;10:51-6.
10) Lund-Andersen H, Kjeldsen CS. Uptake of glucose analogues by rat brain cortex slices:
membrane transport versus metabolism of 2- deoxy-D-glucose. J Neurochem 1977;29:205-11.
11) Owen OE, Morgan AP, Kemp HG, Sullivan JM, Herrera MG, Cahill GF Jr. Brain metabo- lism during fasting. J Clin Invest 1967;46:1589- 95.
12) De Vivo DC. In cerebral metabolism and neural function. 4th ed. Baltimore; Williams and Wilkins, 1980;243-54.
13) De Vivo DC, Leary L, Wang D. Glut-1 defi- ciency syndrome and other glycolytic defects.
J Child Neurol 2002;17:515-23.
14) Magistretti PJ. Cellular bases of functional brain imaging insights from neuron-glia me- tabolic coupling. Brain Res 2000;886:108-12.
15) Vannucci SJ, Maher F, Simpson IA. Glucose transporter proteins in brain delivery of glucose to neuron and glia. Glia 1997;21:2-21.
16) Shows TB, Eddy RL, Byers MG. Polymorphic human glucose transporter gene(GLUT) is on chromosome 1p31.3-p35. Diabetes 1987;36:546-9.
17) Fukumoto H, Seino S, Imura H. Characteriza- tion and expression of human HepG2/erythro- cyte glucose-transporter gene. Diabetes 1988;
37:657-61.
18) Birnbaum MJ, Haspel HC, Rosen OM. Cloning and characterization of a cDNA encoding the rat brain glucose-transporter protein. Proc Natl Acad Sci USA 1986;83:5784-8.
19) Fischbarg J, Kuang K, Vera JC. Glucose transporters serve as water channels. Proc Natl Acad Sci USA. 1990;87:3244-7.
20) Carruthers A. Facilitated diffusion of glucose.
Physiol Rev 1990;70:1135-76.
21) Rumsey SC, Kwon O, Xu GW. Glucose trans- porter isoforms GLUT1 and GLUT3 transport dehydroascorbic acid. J Biol Chem 1997;272:
18982-9.
22) Polt R, Porreca F, Szabo LZ. Glycopeptide enkephalin analogues produce analgesia in mice: evidence for penetration of the blood- brain barrier. Proc Natl Acad Sci USA 1994;
91:7114-8.
23) Agus DB, Gambhir SS, Pardridge WM. Vita- mine C cross the blood-brain barrier in the oxidized form through the glucose transporters.
Clin Invest 1997;100:2842-8.
24) Chugani HT, Phelps ME, Mazziotta JC. Positron emission tomography study of human brain functional development. Ann Neurol 1987;22:
487-97.
25) Wang D, Pascal JM, Yang H, Engelstad K, Jhung S, Sun RP, et al. Glut-1 deficiency syndrome: Clincal, genetic, and therapeutic aspects. Ann Neurol 2005;57:111-8.
26) Hu IC, Singh SP, Snyder AK. Effects of ethanol on glucose transporter expression in cultured hippocampal neurons. Alcohol Clin Exp Res 1995;19:1398-402.
27) Krauss SW, Diamond I, Gordon AS. Selective inhibition by ethanol of the type 1 facilitative glucose transporter(GLUT1). Mol Pharmocol 1994;45:1281-6.
28) Ho YY, Yang H, Klepper J. Glucose transporter type 1 deficiency syndrome(Glut1 DS): me- thylxanthines potentiate GLUT1 haploinsuffi- ciency in vitro. Pediatr Res 2001;50:254-60.
29) Pinkofsky HB, Dwyer DS, Bradley RJ. The inhibition of GLUT1 glucose transport and cytochalasin B binding activity by tricyclic antidepressants. Life Sci 2000;66:271-8.
30) Wang D, Pascal JM, Ho YY. Glut-1 deficiency syndrome(Glut-1 DS): a severe phenotype as- sociated with compound heterozygosity in trans. Ann Neurol 2001;50:S125.
31) Heilig C, Brosius F, Siu B. Implications of glucose transporter protein type 1(GLUT-1)- haplodeficiency in embryonic stem cells for their survival in response to hypoxic stress.
Am J Pathol 2003;163:1873-85.
32) Heilig CW, Saunders T, Brosius FC. Glucose transporter-1-deficient mice exhibit impaired development and deformities that are similar
to diabetic embryopathy. Proc Natl Acad Sci USA 2003;100:15613-8.
33) De Vivo DC, Garcia-Alvarez M, Ronen GM.
Glucose transport protein deficiency: an emer- ging syndrome with therapeutic implications.
Int Pediatr 1995;10:51-6.
34) Pascal JM, Van Heertum RL, Wang D, En- gelstad K, De Vivo DC. Imaging the metabolic footprint of Glut-1 deficiency on the brain.
Ann Neuol 2002;52:458-64.
35) Klepper J, Garcia-Alvarez M, O'Driscoll KR, Parides MK, Wang D, Ho YY, et al. Erythro- cyte 3-O-methyl-D-glucose uptake assay for diagnosis of glucose transporter protein synd- rome. Ann Neurol 1988;44:286-7.
36) Seidner G, Alvarez MG, Yeh JI, O'Driscoll KR, Klepper J, Stump TS, et al. GLUT-1 deficiency syndrome caused by haploinsufficiency of the blood brain barrier hexose carrier. Nat Genet 1998;18:188-91.
37) De Vivo DC, Trifiletti RR, Jacobson RI, Ronen GM, Behmand RA, Harik SI. Defective glucose transport across the blood brain barrier as a cause of persistent hypoglycorrhachia, seizures, and developmental delay. N Engl J Med 1991;
325:703-9.
38) Friedman JR, Thiele EA, Wang D, Levine KB, Cloherty EK, Pfeifer HH, et al. Atypical GLUT1 deficiency with prominent movement disorder responsive to ketogenic diet. Mov Disord 2006;21:241-5.
39) Leary LD, Wang D, Nordli DR Jr. Seizure characterization and electroencephalographic features in Glut-1 deficiency syndrome. Epi- lepsia 2003;44:701-7.
40) Klip A, Tsakiridis T, Marette A, Ortiz PA.
Regulation of expression of glucose transporters by glucose: a review of studies in vivo and in cell cultures. FASEB J 1994;8:43-53.
41) Kulikova-Schupak R, Ho YY, Kranz-Eble P.
Stimulation of GLUT-1 gene transcription by thioctic acid and its potential therapeutic value in Glut-1 deficiency syndrome(Glut-1 DS). J Inherit Metab Dis 2001;24:106.
42) De Vivo DC, Garcia AM, Tristschler HJ. Defi- ciency of glucose transporter protein type 1:
possible therapeutic role for alpha-lipoic acid (thioctic acid). Diabetes und Stoffwechsel 1996;5:36-40.
43) Klepper J, Fischbarg J, Vera JC, Wang D, De Vivo DC. GLUT1-deficiency barbiturates po- tentiate haploinsufficiency in vitro. Pediatr Re 1999;46:677-83.
44) Klepper J, Schaper J, Raca G, Coryell J, Das S, Hayflick SJ, et al. Effects of anticonvulsants on GLUT1-mediated glucose transport in GLUT1 deficiency syndrome in vitro. Eur J Pediatr 2003;162:84-9.
45) Brockmann K, Wang D, Korenke CG, von Moers A, Ho YY, Pascual JM, et al. Autoso- mal dominant glut-1 deficiency syndrome and familial epilepsy. Ann Neurol 2001;50:476-85.
46) Guillam MT, Hummler E, Schaerer E, Yeh JI, Birnbaum MJ, Beermann F, et al. Early dia- betes and abnormal postnatal pancreatic islet development in mice lacking Glut-2. Nat Genet 1997;17:327-30.
47) Stenbit AE, Tsao TS, Li J, Burcelin R, Geenen DL, Factor SM, et al. GLUT4 heterozygous knockout mice develop muscle insulin resistance and diabetes. Nat Med 1997;3:1096-101.
48) Wang D, Pascual JM, Yang H, Engelstad K, Moa X, Cheng J, et al. A mouse model for glut-1 haploinsufficiency. Human Mol Genet 2006;15:1169-79.