저작자표시-비영리-변경금지 2.0 대한민국 이용자는 아래의 조건을 따르는 경우에 한하여 자유롭게 l 이 저작물을 복제, 배포, 전송, 전시, 공연 및 방송할 수 있습니다. 다음과 같은 조건을 따라야 합니다: l 귀하는, 이 저작물의 재이용이나 배포의 경우, 이 저작물에 적용된 이용허락조건 을 명확하게 나타내어야 합니다. l 저작권자로부터 별도의 허가를 받으면 이러한 조건들은 적용되지 않습니다. 저작권법에 따른 이용자의 권리는 위의 내용에 의하여 영향을 받지 않습니다. 이것은 이용허락규약(Legal Code)을 이해하기 쉽게 요약한 것입니다. Disclaimer 저작자표시. 귀하는 원저작자를 표시하여야 합니다. 비영리. 귀하는 이 저작물을 영리 목적으로 이용할 수 없습니다. 변경금지. 귀하는 이 저작물을 개작, 변형 또는 가공할 수 없습니다.
The Role of Connexin43 and Gap Junctional
Intercellular Communication in the Apoptotic
Process of Auditory Cells
by
Yeon Ju Kim
Major in Molecular Medicne
Department of Biomedical Sciences
The Graduate School, Ajou University
The Role of Connexin43 and Gap Junctional
Intercellular Communication in the Apoptotic
Process of Auditory Cells
by
Yeon Ju Kim
A Dissertation Submitted to The Graduate School of
Ajou University in Partial Fulfillment of the Requirements for
The Degree of Master of Biomedical Sciences
Supervised by
Yun-Hoon Choung, M.D., D.D.S., Ph.D.
Major in Molecular Medicine
Department of Biomedical Sciences
The Graduate School, Ajou University
- ABSTRACT -
The Role of Connexin43 and Gap junctional Intercellular
Communication in the Apoptotic Process of Auditory Cells
Cis-diamminedichloroplatinum (cisplatin) is one of effective chemotherapeutic drugs
for cancer therapy. Nevertheless, most patients treated with cisplatin are exposed to high risk of ototoxicity such as hearing loss or dizziness. It has been reported that ototoxicity occurs in about 75-100 % of patients treated with cisplatin and shows irreversible, cumulative and bilateral characteristics. However, cisplatin-induced ototoxic mechanisms have not been clearly identified yet even through many studies. Histochemical studies on the distribution of cisplatin have shown that spiral ganglions, lateral wall cells, and organ of Cortis are major targets, where many connexins (Cxs) are expressed. Cxs are a family of membrane proteins and these are expressed abundantly in mammalian inner ears. Cxs oligomerize into a hexameric connexon, also known as a hemichannel, and transport to plasma membranes along microtubules finally forming a gap junction between cells. In the inner ear, gap junctions play a critical role in the generation the endocochlear potential, which is required for normal hearing.
The aim of this study was to determine the role of gap junctions and their constituents, hemichannels and Cxs in the cisplatin-induced ototoxic mechanism. As a result, I found that inhibition of gap junctional activity with either Cx43 siRNA transfection or a pharmacological drug, 18α-glycyrrhetinic acid in HEI-OC1 auditory cells caused a marked increase in cell viability with a decrease of apoptosis based on cleaved Poly (ADP-ribose)
polymerase (PARP) and cleaved caspase 3 levels. Furthermore, I demonstrated that extracellular signal-regulated kinase (ERK) and protein kinase B (Akt) pathways had a significant role in preventing cell death by inhibition of gap junctions. Next, I found that cisplatin did not affect the opening and closure of Cx43-hemichannels in HEI-OC1 cells and Cx43-transfected Cx-deficient HeLa cells. Accumulation of Cx43 was observed around the nuclei of cisplatin-treated cells, whereas Cx43 was scattered in the punctuated form in the cytoplasms as well as on the membranes in normal cells, suggesting that cisplatin may interrupt the trafficking of Cx43s to cell membranes in HEI-OC1 cells. In addition, I investigated whether Cx43 protein itself affected cell viability in response to cisplatin. I found that the knockdown of Cx43 in HEI-OC1 cells resulted in a significant increase in cell viability following cisplatin treatment independent of gap junctions. This response did not change following inhibition of Cx43 transport to the plasma membrane by Brefeldin A. HeLa cells expressing either full lengthen Cx43 and its N-terminal domain or its carboxyl tail, showed higher sensitivity to cisplatin than mock-treated cells. The mitochondrial Cx43 protein level was significantly increased in HEI-OC1 cells after cisplatin treatment in a time-dependent manner, which coincided with the release of mitochondrial cytochrome C into the cytoplasm.
These findings suggest that Cx43 plays important roles on cisplatin-induced ototoxic damage through gap junction-dependent or independent processes in auditory cells. The control of the Cx43-mediated signaling may be a potential target for therapeutic strategies for drug-induced ototoxicity.
TABLE OF CONTENTS
ABTRACT ··· i
TABLE OF CONTENTS ··· iii
LIST OF FIGURES ··· v
. Ⅰ INTRODUCTION ··· 1
. Ⅱ MATERIALS AND METHODS ··· 8
1. Cell culture ··· 8
2. Plasmid constructs and transfection ··· 8
3. Fluorescence Recovery After Photobleaching (FRAP) ··· 9
4. Scrape Load Dye Transfer (SLDT) Technique ··· 10
5. Lucifer yellow dye uptake ··· 11
6. Live/Dead assay ··· 11 7. MTS assay ··· 11 8. Western blot ··· 11 9. Immunocytochemistry ··· 12 10. Immunohistochemistry ··· 13 11. Animals ··· 13 12. Animal study ··· 14
13. Scanning Electron Microscopy (SEM) ··· 14
14. Auditory brainstem response (ABR) test ··· 16
15. Statistical analysis ··· 16
. Ⅲ RESULTS ··· 17
1. Auditory cells expressed functional gap junctions ··· 17 2. Gap junction activity was interrupted by cisplatin ··· 22 3. Inhibition of gap junctions reduced cisplatin-induced apoptosis ··· 27 4. Activation of ERK and Akt contributed to inhibition of gap junction-mediated
beneficial effects in response to cisplatin ··· 33 5. Design of gap junctional and non-junctional condition in auditory cell culture 38 6. Effects of gap junction inhibition on cisplatin toxicity in the junctional and
non-junctional condition ··· 41 7. Cx43 hemichannels was not changed by cisplatin treatment ··· 43 8. HeLa cells transfected with cytoplasmic CT domain of Cx43 become sensitive
to the cisplatin ··· 48 9. Cisplatin-induced toxicity decreases following Cx43 knockdown by inhibition
of Cx trafficking to the plasma membrane ··· 51 10. Mitochondrial Cx43 has an impact on the cisplatin-induced damage in
HEI-OC1 cells ··· 55 11. Phosphorylation status of Cx43 in primary organ of Corti cells treated with
gap junction blocker ··· 57 12. Effects of CBX and 18α-GA on the cisplatin-induced hearing loss ··· 59 . Ⅳ DISCUSSION ··· 62 . Ⅴ CONCLUSION ··· 70 REFERENCES ··· 71 국문요약 ··· 82
LIST OF FIGURES
Fig. 1. Transmission electron microscope images of gap junctions between rat
cochlear supporting cells ··· 5
Fig. 2. Gap junction structure ··· 6
Fig. 3. Diagram of the cochlea cellular systems showing the gap junction networks ··· 7
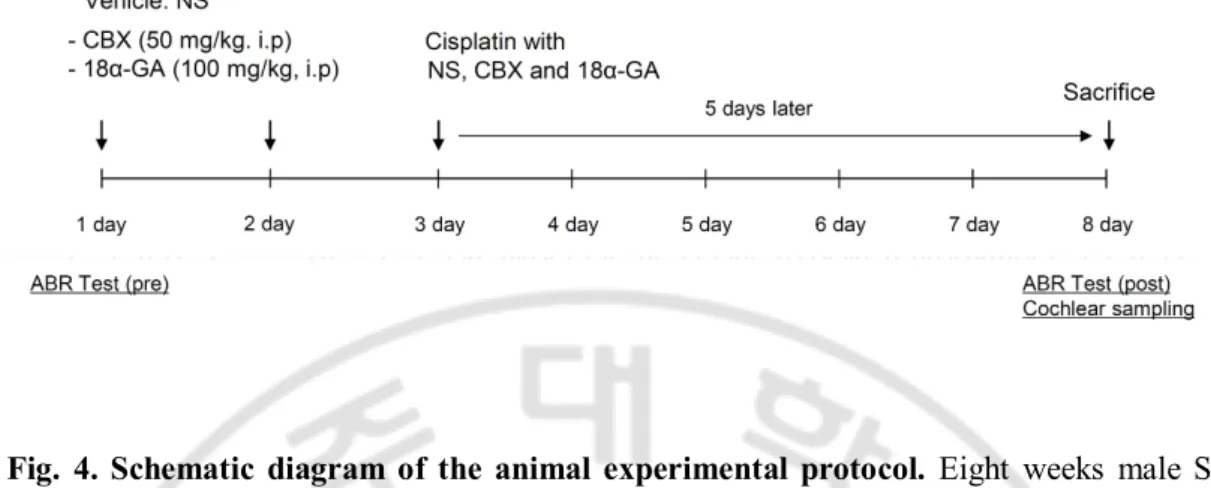
Fig. 4. Schematic diagram of the animal experimental protocol ··· 15
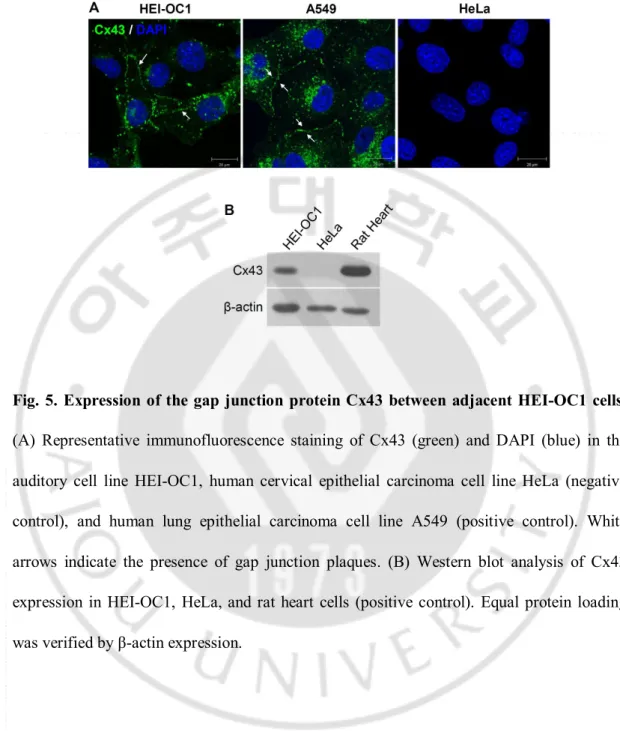
Fig. 5. Expression of the gap junction protein Cx43 between adjacent HEI-OC1 cells ··· 19
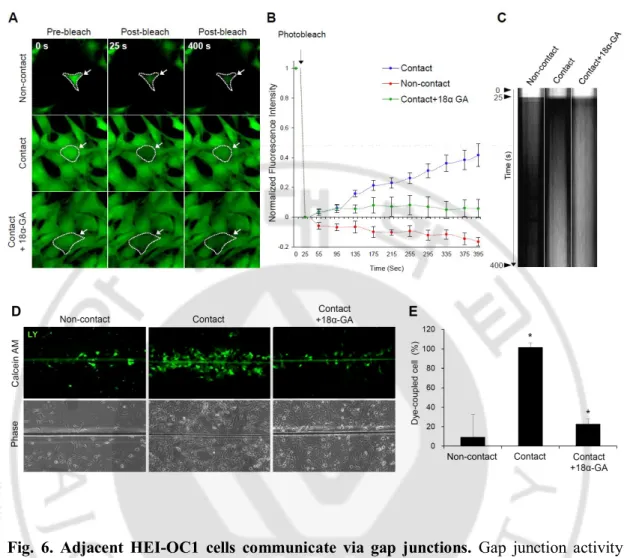
Fig. 6. Adjacent HEI-OC1 cells communicate via gap junctions ··· 20
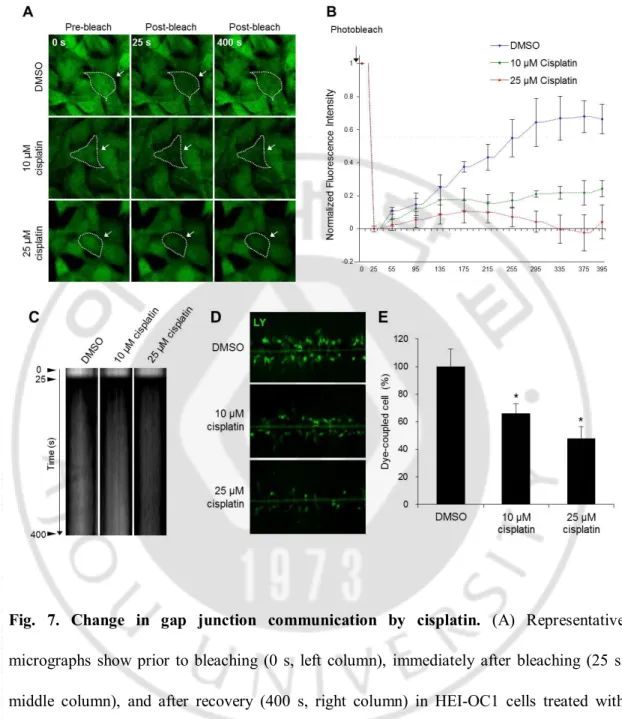
Fig. 7. Change in gap junction communication by cisplatin ··· 24
Fig. 8. Cisplatin inhibits the trafficking of Cx43 to cell membranes ··· 26
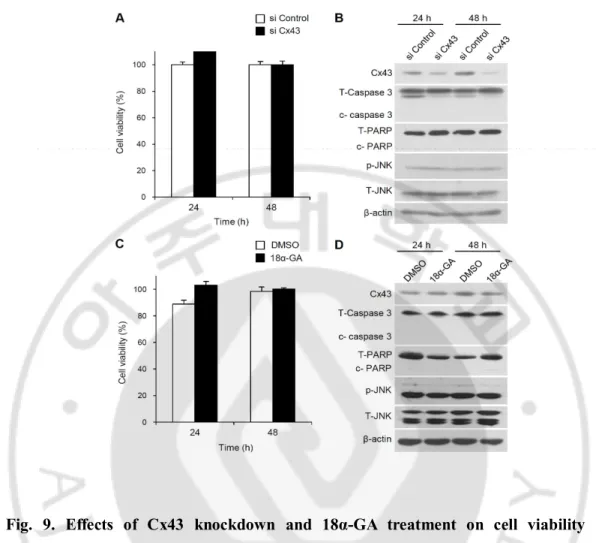
Fig. 9. Effects of Cx43 knockdown and 18α-GA treatment on cell viability and activation of caspase 3, PARP, and JNK activation ··· 29
Fig. 10. Effects of blocking gap junction intercellular communication on cisplatin -induced HEI-OC1 cell death ··· 30
Fig. 11. Quantitative analysis of photobleaching (FRAP) assay for gap junction
activity ··· 32
Fig. 12. Effects of blocking gap junction intercellular communication on cisplatin
-induced MAPK activation ··· 35
Fig. 13. Knockdown of Cx43 and gap junction blocker-induced increase in
cell viability was attenuated by ERK and Akt inhibition ··· 37
Fig. 14. Junctional and non-junctional Cx43s in cell density-dependent cultures ··· 39
Fig. 15. Effect of 18α-GA and Cx43 knockdown on cisplatin cytotoxicity in the
junctional and non-junctional condition ··· 42
Fig. 16. Change of hemichannel function in HEI-OC1 cells by cisplatin treatment ··· 45
Fig. 17. Altered hemichannel function following cisplatin treatment in Cx43-
transfected HeLa cells ··· 46
Fig. 19. Effect of cisplatin on cell viability in HeLa cells expressing Cx43-FL, NT
and CT ··· 50
Fig. 20. Effect of Cx43 knockdown on cisplatin-induced cytotoxicity in HEI-OC1
cells in the non-junctional region by BFA ··· 53
Fig. 21. Change in mitochondrial Cx43 during cisplatin-induced cytotoxicity
in HEI-OC1 cells ··· 56
Fig. 22. The expression of Cx43 in the cochlea and primary organ of Corti cells··· 58
Fig. 23. The effect of gap junction blockers (CBX and 18α-GA) in cisplatin
-induced hearing loss on auditory brainstem response ··· 60
I. INTRODUCTION
Cis-diamminedichloroplatinum (cisplatin) is an effective chemotherapeutic drug
that has been widely used to treat various types of cancers (Rybak et al., 2007). However, its clinical use is quite limited due to its severe side effects such as ototoxicity and nephrotoxicity (Rybak et al., 2009). In particular, ototoxicity has been reported to occur in about 75–100% of patients treated with cisplatin and has been shown to have irreversible, cumulative, and bilateral characteristics (McKeage, 1995). However, the mechanism mediating cisplatin-induced ototoxicity has not been clearly elucidated. Histochemical studies on the effects of cisplatin have shown that hair cells and supporting cells in the organ of Corti are major targets, and exposure of these cells to cisplatin leads to cell death or dysfunction (Rybak et al., 2007). Cisplatin has been reported to accumulate in the spiral ganglion and the lateral wall, including spiral ligaments and stria vascularis (Campbell et al., 1999; Li et al., 2006). Cisplatin is transferred into hair cells through several transporters, i.e., copper transporter 1 (More et al., 2010) and organic cation `transporter 2 (Ciarimboli et al., 2010), and causes increases in the expression of NADPH oxidase 3 (NOX3) and the superoxide generating enzyme (Banfi et al., 2004; Mukherjea et al., 2010). With the rise in NOX3, large amounts of reactive oxygen species (ROS) are produced (Rybak et al., 2009), subsequently activating the mitogen-activated protein kinases (MAPK) pathway and promoting the release of cytochrome C from mitochondria (Devarajan et al., 2002; Mukherjea and Rybak, 2011); this pathway can lead to apoptosis via caspase activation (Lee et al., 2010). Despite these previous reports, the mechanisms underlying cisplatin-induced ototoxicity are not yet fully understood.
Gap junctions are one of the most important pathways involved in intercellular communication in the cochlea (Martinez et al., 2009) (Fig. 1). Gap junctions permit the direct intercellular transfer of molecules less than 1 kilodalton (kDa). Each gap junction is formed by the apposition of two hemichannels (or connexon) from adjacent cells, and each in turn is composed of six protein subunits termed connexin (Cx) (Zampighi, 1980). The Cx consists of four transmembrane domains, two extracellular loops, a cytoplasmic loop, a cytoplasmic amino-terminal (NT), and a carboxy-terminal (CT) domain (Segretain and Falk, 2004). To date, the Cx family comprises twenty-one members in the human and twenty in the mouse, and these are expressed in a tissue-specific manner (Martin et al., 1998).
In general, gap junctional intercellular communication is thought to play important roles in cellular homeostasis, embryonic development, proliferation, differentiation, growth, and physiological modulation. Recent studies have suggested that gap junctions are also critical regulators under pathological conditions (Garcia-Dorado et al., 2004; Krutovskikh et al., 2002). In this regard, some studies have shown that gap junction coupling is controlled by injurious agents, which, in turn, can either weaken or worsen the damage, the so-called “Good Samaritan effect” and “Bystander effect”, respectively (Farahani et al., 2005). The “Bystander effect” has been particularly well documented in cancers (Carrio et al., 2001). Cxs are degraded in most cancers, and the upregulation of gap junctional communication and overexpression of Cxs have been shown to potentiate anticancer drug efficacy and to reduce drug resistance (Garcia-Rodriguez et al., 2011; Sun et al., 2012a). In contrast, neuronal cells are more susceptible to toxic exposure or focal ischemia by gap junction blocking agents, suggesting that gap junctions provide a “Good Samaritan effect” under conditions that promote cytotoxicity (Blanc et al., 1998). Thus, the role of gap junctions may differ
according to the cell type, period, and severity of the injury (Ahmad et al., 2003; Perez Velazquez et al., 2003). Recently, there is ample evidence suggesting that non-junctional hemichannels can also be opened or closed in several pathological conditions without gap junction formation. This in turn allows the exchange of molecules lower than 1 kDa in size, comparable to the action of gap junctions (Decrock et al., 2009). In addition, several studies using gene deletion mutants to study channel formation reported that the Cx protein itself could affect cellular function in a junction-independent manner (Moorby and Patel, 2001). Therefore, there is a need for a comprehensive understanding of Cx in pathological situations, including an important role in cisplatin-induced ototoxicity.
In the mammalian inner ear, gap junctions are abundant in the cochlear and vestibular parts, where Cx26, Cx30, Cx31 and Cx43 are expressed (Ahmad et al., 2003; Suzuki et al., 2003). These gap junctions play a critical role in the circulation of K+ between
the fluids of the inner ear (Zdebik et al., 2009). The circulation of K+ is also essential for the
maintenance of specialized ionic composition and positive endocochlear potential of 80 to 100 mV in endolymph, which are required for normal hearing (Kikuchi et al., 2000a; Kikuchi et al., 2000b) (Fig. 2). Furthermore, it has been suggested that gap junctions are involved in Ca2+ signaling and energy metabolism through controlling the passage of small
molecules such as inositol trisphosphate, adenosine triphosphate (ATP), cyclic adenosine monophosphate (cAMP) and cyclic guanosine monophosphate (cGMP) (Chang et al., 2008; Zhao et al., 2006). Mutations in human Cx26, Cx30, Cx31 and, Cx43 have been linked to syndromic and nonsyndromic deafness. It has been demonstrated that the knockout mouse of Cx genes in the ear caused severe hearing loss (Kudo et al., 2003; Teubner et al., 2003; Zhao et al., 2006). Together, these findings indicate that inner ear gap junctions may be very
crucial for normal hearing system.
I propose herein that both gap junctional intercellular communication and non-juntional Cxs may play critical roles ototoxic-dug induced hearing loss. Therefore, I investigated the effect of cisplatin on junctional and non-junctional Cxs in auditory cells using in vitro and in vivo experiments.
Fig. 1. Transmission electron microscope images of gap junctions between rat cochlear supporting cells. White arrows indicate the presence of gap junction plaques.
Fig. 2. Gap junction structure. (A) Six connexin monomers oligomerize into connexons or
hemichannels, which combine into gap junction channels. (B) The connexin protein creates four membrane domains (M1-M4) with two extracellular loops (E1 and E2), one cytoplasmic loop (C), and the N and C termini exposed to the cytoplasm.
Fig. 3. Diagram of the cochlea cellular system showing gap junction networks. Deiter
cells (pink) and supporting and epithelial cells (light pink) constitute the epithelial gap junction network. Fibrocytes and stria vascularis (orange) constitute the connective tissue gap junction network. It has been proposed that K+ recirculates back to the endolymph
II. MATERIALS AND METHODS
1. Cell culture
House Ear Institute-Organ of Corti (HEI-OC1), a conditionally immortalized cochlear epithelial cells and human cervical carcinoma cells (HeLa) were grown in Dulbecco’s Modified Eagles Media (DMEM; Gibco-BRL, Grand Island, NY, USA) supplement with 10 % fetal bovine serum (FBS; Sigma-Aldrich; St. Louis, MO, USA). The HEI-OC1 cells were incubated at 33 °C in a humidified atmosphere containing 10 % CO2
because it harbors temperature-sensitive large T antigen (Jat et al., 1991) and HeLa cells were incubated at 37 °C in a 5 % CO2. Cisplatin (Sigma-Aldrich, USA) were used for the
induction of ototoxicity. The gap junction inhibitor, 18 alpha-glycyrrhetinic acid (18α-GA; Sigma-Aldrich, Germany) and carbenoxolone (CBX; Sigma-Aldrich, Sigma-Aldrich; St. Louis, MO, USA) were used to block gap junction communication between the cells. The brefeldin A (BFA; Sigma-Aldrich; St. Louis, MO, USA) was used to inhibit assembly of gap junction. To explore the effects of mitogen-activated protein kinase (MAPK) inhibition, 10 μM LY294002 (a phosphoinositide 3-kinase [PI3K] inhibitor) and 20 μM PD98059 (a MAPK/extracellular signal-regulated kinase [ERK] inhibitor) were added to the media 2 h prior to treatment with cisplatin. All chemicals for MAPK inhibition were supplied by Calbiochem (San Diego, CA, USA).
2. Plasmid constructs and transfection
Reverse transcription polymerase chain reaction (RT-PCR) was performed using standard protocols. Primer sequences for the full length of Cx43 (Cx43-FL) were
5′-TTAGGCTAGCATGGGTGACTGGAGCGCCTTAGG-3′ (Forward primer),
5′-TTAGGCTAGCATGGGTGACTGGAGCGCCTTAGG-3′ (Reverse primer). After
amplification of human cDNA, the Cx43 product was cloned into pcDNA 3.1 (-) (5427 bp) (Invitrogen, OR, USA) using NheI and EcoRV restriction enzyme sites according to the manufacturer’s instructions. The cDNA fragment encoding the N-terminal (Cx43-NT, amino acid 1-256) and carboxyl terminal domains (Cx43-CT, amino acid 243-382) were synthesized by PCR using human Cx43 cDNA and were then cloned into the pcDNA 3.1 (-) plasmid using XbaI and EcoRV restriction enzymes. Primer sequences for each domain were as follows: CX43-NT: 5′-TTAGTCTAGAATGGAGCGACCCTTACCATGCGA-3′ (Forward primer), 5′-ACATGATATCCTAGATCTCCAGGTCATCAGGC-3′ (Reverse primer) and Cx43-CT: TTAGTCTAGAATGGGTGACTGGAGCGCCTTAG-3′ (Forward primer), 5′-ACATGATATCCTAGGGCTCAGCGCACCACTGGTC-3′ (Reverse primer). Plasmid DNA was prepared using a plasmid mini preparation kit (Solgent, Daejeon, Korea) and plasmid constructs were verified by Sanger sequencing (Macrogen, Korea). The empty pcDNA3.1 (-) construct was used as an empty vector control. To silence the expression of Cx43, short interfering RNA (siRNA) was used (Santa Cruz Biotechnology, Santa Cruz, CA). A scrambled siRNA oligo was used as the control. Lipofectamine 2000 or RNAi MAX was used for all transfections (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions.
3. Fluorescence Recovery After Photobleaching (FRAP)
Monolayer HEI-OC1 cells were washed with PBS, and calcein AM (623 Da, Molecular Probes, Eugene, OR) in 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
(HEPES) was loaded for 15 min. Dye-loaded cells were placed in an incubation chamber (21°C) under a Zeiss LSM 700 confocal microscope (Carl Zeiss MicroImaging Inc.). To test gap junctions, I selected one cell that was adjacent to least 3 other cells and photobleached the cell with a short intense exposure to an argon laser. After photobleaching, the recovery of fluorescence was monitored every 10 s for 400 s. The fluorescence intensity of the bleached region was measured using LSM confocal software. The percent recovery was calculated according to the following formula (Chi et al., 2005): Recovery (%) = (Ft – F0/Fb) × 100 (Ft:
fluorescence at each time point after photobleaching, F0: fluorescence at 0 s after photobleaching, Fb: fluorescence before photobleaching). To compare recovery (%) between
cells, I normalized the fluorescence intensity of postbleaching to 0. Additionally, to assess tendencies of fluorescence kinetics in an image time series, I analyzed images by kymography using METAMORPH software (Universal Imaging Corp., Downington, PA, USA). I defined a horizontal rectangular region of interest (ROI), and intensities were measured within each image.
4. Scrape Load Dye Transfer (SLDT) Technique
HEI-OC1 cells were plated and grown overnight to confluence in 35-mm culture dishes. Next, 0.05% Lucifer yellow (LY; Sigma-Aldrich) in PBS was added to cover the cells, and cells were randomly scraped with a sharp blade. After incubating for 5 min at 33°C, cells were washed with PBS and fixed with 4 % paraformaldehyde. Dye-coupled cell layers were observed using a fluorescence microscope. The numbers of dye-coupled cells were counted to evaluate gap junction intercellular communication.
5. Lucifer yellow dye uptake
HEI-OC1 cells, HeLa cells transfected with Cx43 or empty vector were incubated with 2 mM LY in full nutrient media, Ca2+ free media, 100 μM cisplatin, and 1mM H
2O2
containing media. To evaluate the effect of hemichannel blockers, cells were pretreated with 18α-GA or CBX for 2 h and LY uptake was measured by an Axiovision inverted microscope (Carl Zeiss AG, Gottingen, Germany)
6. Live/Dead assay
Cell viability assays were performed according to the manufacturer’s protocol (Invitrogen Molecular. Probes, Carlsbad, CA). Briefly, cells were washed with PBS, and a mixed solution of calcein AM and ethidium homodimer-1 (EthD-1) was added to cells. After incubation for 30 min at room temperature, cells were imaged under an Axiovision fluorescence microscope (Carl Zeiss AG, Gottingen, Germany).
7. MTS assay
Cells were seeded in a 96-well microtiter plate and allowed to adhere for 24 h before treatment with cisplatin ± agents, following which 20 μM of MTS was added to each well. The plate was incubated for 4 h at 33°C and then absorbance (490 nm) was measured using a microplate reader (Bio-Rad Model 680, Hercules, CA, USA). All assays were repeated at least three separate times.
Cells were washed with phosphate-buffered saline (PBS) and lysed using radio-immunoprecipitation assay (RIPA) buffer. Mitochondrial and cytosolic fractions were extracted using a mitochondrial isolation kit (Pierce, Rockford, IL) in accordance with the manufacturer's instructions. Equal amounts of protein from each sample were analyzed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) followed by electro-transfer to polyvinylidene fluoride (PVDF; Millipore, Billerica, MA, USA) membranes. Membranes were blocked for 1 h with 5% skim milk in PBS containing 0.05% Tween20 (PBST) and incubated with primary antibodies overnight at 4°C. Membranes were washed 4 times with PBST and incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies for 1 h at room temperature. After washing, the protein signal was detected using a chemiluminescence solution (GenDEPOT, Barker, TX, USA). The intensity of protein bands was quantified using Image J software (Broken Symmetry Software, USA). The following antibodies were used: anti-Connexin 43, anti-PARP, anti-phospho-p44/42 MAPK, anti-total-p44/42 MAPK, anti-caspase 3, anti-phospho-Akt, anti-phospho-SAPK/c-Jun N-terminal kinase (JNK), anti-total-SAPK/JNK, anti-COX IV (Cell Signaling, Danvers, MA, USA), α-tubulin (Santa Cruz biotechnology; Santa Cruz, CA, USA) and anti-cytochrome C (Enzo Life Sciences, Lorrach, Germany). β-Actin was used as a loading control.
9. Immunocytochemistry
For immunocytochemical analysis, HEI-OC1 cells were grown on glass coverslips (Marienfeld, Lauda-Koenigshofen, Germany). Cells were fixed in 4% paraformaldehyde for 20 min, washed with PBS, and permeabilized in 0.2% Triton-X 100/PBS for 10 min at room
temperature. After washing, they were blocked with 1% bovine serum albumin (BSA; GenDEPOT) in PBS and processed for indirect immunofluorescence using primary antibodies and secondary antibodies coupled with fluorescein isothiocyanate (FITC) or cyanine 3 (Cy3) (Jackson ImmunoResearch Laboratories, West Grove, PA, USA). The primary and secondary antibodies were diluted in a blocking buffer. Nuclei were stained with 1 μg/mL 4′, 6′-diamidino-2-phenylindole (DAPI; Invitrogen) in PBS for 2 min at room temperature. The cells were visualized with a Zeiss LSM 700 confocal or AxioVision LE 4.5 microscope (Carl Zeiss MicroImaging Inc., Thornwood, NY, USA).
10. Immunohistochemistry
Frozen sections were dried at 37℃ for 1 h. After permeabilized with 0.2 % Triton X-100, blocked with 1 % BSA for 30 min, and sections were incubated with antibodies at 4 ℃ for 24 h. Following washing with PBS, the sections were incubated with FITC or Cy3-conjugated secondary antibodies at room temperature for 1 h. After washing, the sections were mounted with mounting medium with DAPI (Vector Laboratories, Burlingame, CA, USA).
11. Animals
Frozen sections were dried at 37℃ for 1 h. After permeabilized with 0.2 % Triton X-100, blocked with 1 % BSA for 30 min, and sections were incubated with antibodies at 4 ℃ for 24 h. Following washing with PBS, the sections were incubated with FITC or Cy3-conjugated secondary antibodies at room temperature for 1 h. After washing, the sections were mounted with mounting medium with DAPI (Vector Laboratories, Burlingame, CA,
USA).
12. Animal study
Male Sprague-Dawley rats (8 weeks, 230-270 g) were housed in a temperature-controlled (23 ± 2 ℃) room with a 12 h light/dark cycle. The rats were provided with free access to standard laboratory food and tap water. Sixteen rats were divided into 4 groups (n=4), normal saline, cisplatin, cisplatin + CBX and cisplatin + 18α-GA group. Cisplatin was dissolved in saline at a concentration of 0.15 mg/ml, and was administered intraperitoneally (i.p) at a dose of 16 mg/kg. For cisplatin+CBX and cisplatin+18α-GA, CBX (50 mg/kg) and 18α-GA (100 mg/kg) was intraperitoneally pre- and co- injected to rats treated with cisplatin. Hearing function was evaluated before and after drug injection. Cochlea was harvested for morphological evaluation (Fig. 4).
13. Scanning Electron Microscopy (SEM)
Rats were decapitated quickly after being anesthetized, temporal bones harvested. The perilymphatic space was perfused with 4 % gluteraldehyde. Each specimen was then placed in the same gluteraldehyde solution overnight at 4℃. After rinsing 3 times with phosphate-buffered saline (PBS), the end-organ surfaces were prepared for SEM. Samples were serially dehydrated in 50 %, 70 %, 90 %, 95 %, and 100 % acetone. Each specimen was exposed to hexamethyldisilazane and allowed to air dry, then placed on a stub for gold-sputter coating. Photographs were taken with a JSM-5410 LV SEM camera (Jeol, Tokyo, Japan). The number of hair cells in the organ of Corti was analyzed as previously described (Choung., 2011).
Fig. 4. Schematic diagram of the animal experimental protocol. Eight weeks male SD
rats were pre-injected by the 50 mg/kg carbenoxolone (CBX) and 100 mg/kg 18α-GA once every other day. On day 3, rats were injected with 16 mg/kg cisplatin with normal saline (NS), CBX and 18α-GA. Hearing function was assessed 2 days before and 5 days after drug injection. On day 8, all rats were sacrificed and cochleas were harvested for morphological evaluation.
14. Auditory brainstem response (ABR) test.
The auditory brainstem response (ABR) was tested with the Biosig 32 system (Tucker-Davis Technologies, Gainesville, FL, USA) as previously described. (Choung et al., 2011) The threshold was defined as the lowest stimulus level at which a clear waveform was visible in the evoked trace.
15. Statistical analysis
All values are expressed as mean (± standard deviation; s.d.). Statistical significance between the groups was calculated by the Mann-Whitney U test using SPSS software version 12.0 (SPSS Inc., Chicago, Illinois, USA). Probability values (P-value) less than 0.05 were considered statistically significant.
III. RESULTS
1. Auditory cells expressed functional gap junctions
The mouse organ of Corti-derived HEI-OC1 cell line expresses Cxs, and was used as an in vitro model to study ototoxicity (Decrock et al., 2009). To investigate whether Cx43 was expressed in these cells and whether gap junction channels could form to facilitate communication between adjacent cells, HEI-OC1 cells were grown to 90% confluence on cover slips. As shown in Fig. 5A, I observed diffuse Cx43 expression in the cytoplasm and punctuate Cx43 expression in the plasma membranes between HEI-OC1 cells. Western blot analysis also revealed that HEI-OC1 cells expressed Cx43 (Fig. 5B). In order to assess the function of gap junctions in HEI-OC1 cells, I used 2 parallel techniques: FRAP and SLDT assay. To test whether gap junctions could mediate intercellular communication, I compared contact cells with noncontact sequestered HEI-OC1 cells. HEI-OC1 cells were incubated in calcein AM, which passes readily through gap junctions. The cells were photobleached by exposure to about 5 s of intense light from an argon laser. The recovery of fluorescence intensity was monitored every 10 s for 400 s. As shown in Fig. 6A and B, contact with adjacent HEI-OC1 cells permitted recovery of about 42% of the prebleach signal 400 s after photobleaching, whereas noncontact sequestered HEI-OC1 cells tended to exhibit a decrease in the rate of recovery by 16%. In addition, I observed that the rate of recovery in HEI-OC1 cells was decreased by 36% in cells treated with 18 α-GA, a gap junction blocker, in comparison with DMSO-control cells. To represent the discrimination of fluorescence kinetics during recovery after photobleaching, a kymographic analysis was performed as described in Fig. 6C. Following photobleaching, fluorescence signal appeared gradually in
contact HEI-OC1 cells. In contrast, noncontact sequestered HEI-OC1 cells exhibited reduced fluorescence signals, and no changes were observed in 18α-GA-treated contact HEI-OC1 cells. To investigate whether there was movement of molecules in HEI-OC1 cells facilitated by connexin, I compared HEI-OC1 cells with the connexin-deficient human cancer cell line, HeLa (Lampe et al., 1991). The rate of fluorescence recovery in HeLa cells was significantly lower than in HEI-OC1 cells (data not shown). I observed similar results by SLDT assay in HEI-OC1 cells. The number of dye-coupled cells decreased by 91.6% ± 5.2% at low density and 78.2% ± 4.4% under treatment with 18 α-GA compared to DMSO-treated high-density cells (Fig. 6D and E). Together, these data demonstrated that the gap junction protein Cx43 was expressed abundantly in HEI-OC1 cells and communicate with each other through functional gap junctions.
Fig. 5. Expression of the gap junction protein Cx43 between adjacent HEI-OC1 cells.
(A) Representative immunofluorescence staining of Cx43 (green) and DAPI (blue) in the auditory cell line HEI-OC1, human cervical epithelial carcinoma cell line HeLa (negative control), and human lung epithelial carcinoma cell line A549 (positive control). White arrows indicate the presence of gap junction plaques. (B) Western blot analysis of Cx43 expression in HEI-OC1, HeLa, and rat heart cells (positive control). Equal protein loading was verified by β-actin expression.
Fig. 6. Adjacent HEI-OC1 cells communicate via gap junctions. Gap junction activity
was measured by either fluorescence recovery after photobleaching (FRAP) assay (A-C) or scrape loading dye transfer (SLDT) assay (D,E). (A) HEI-OC1 cell monolayers were incubated in calcein AM (2 μM, 10 min, green) and photobleached with a laser. Recovery of fluorescence in the photobleached cell was monitored every 10 s for 400 s. The region of bleaching is indicated by dotted lines with arrows. Representative confocal micrographs obtained prior to beaching (0 s, left panel), immediately after bleaching (25 s, middle panel), and after recovery (400 s, right panel) in HEI-OC1 cells in noncontact, contact, and treated with the gap junction inhibitor, 18α-GA (25 μM, 2 h). Scale bar = 20 μm. (B) Summary of time course of FRAP data for many individual cells. For every bleached cell, the
fluorescence signals present in each cell immediately prior to and immediately after photobleaching were normalized to 100% and 0%, respectively. (C) Kymograph analysis of the movement of calcein AM dyes in HEI-OC1 cells in noncontact, contact, and treated with the gap junction inhibitor, 18 α-GA. Time-lapse images were collected at 10-s intervals before and after photobleaching in the selected ROI. The horizontal lines of the kymograph represent the time of the image series. (D) Lucifer yellow was loaded by scrape line in monolayers of HEI-OC1 cells in noncontact, contact, and treated with the gap junction inhibitor, 18α-GA (25 μM, 2 h). Cells were monitored by fluorescence (lower) and phase-contrast (upper) microscopy. (E) A bar graph shows the percentage of dye-coupled cells in contact, noncontact, and the group treated with the gap junction inhibitor, 18α-GA. Data are presented as the mean ± SD, P < 0.05; n ≥ 3 independent experiments
2. Gap junction activity was interrupted by cisplatin
I evaluated the cytotoxicity of cisplatin (10–50 μM) in HEI-OC1 cells by MTS assay. After 24 h of incubation with cisplatin (10, 25, or 50 μM), cell viability was decreased significantly by 15.8% ± 5.6%, 39.8% ± 7.3%, and 73.0% ± 8.3%, respectively, compared to DMSO-treated control cells (data not shown). At the molecular level, protein bands representing cleaved caspase 3, cleaved PARP, and phosphorylated-JNK (p-JNK) were increased in cisplatin-treated cells in a dose-dependent manner compared to DMSO-treated control cells (data not shown). To examine whether gap junctions could be affected by cisplatin, I cultured HEI-OC1 cells in the presence of 10 or 25 μM cisplatin for 24 h. I observed that cisplatin blocked intercellular gap junction communication. At 400 s after photobleaching, the rates of recovery for 0, 10, and 25 μM cisplatin were 66.4% ± 0.9%, 24.3% ± 0.5%, and 4.1% ± 1.0%, respectively (Fig. 7A). Kymographic analysis showed a gradual appearance of fluorescence intensity after photobleaching both in DMSO-treated and 10 μM cisplatin-treated HEI-OC1 cells; however, the fluorescence intensity did not change in 25 μM cisplatin-treated HEI-OC1 cells during time-lapse recording (Fig. 7B). In the SLDT assay, decreased numbers of dye-coupled cells were observed for 10 μM (34.1% ± 6.9%) and 25 μM (52.4% ± 8.9%) cisplatin, respectively, when compared to that of DMSO-treated control cells (100% ± 12.8%; Fig. 7D, E). Since I showed that functional gap junctions were decreased by cisplatin, I next considered the expression levels of Cxs in HEI-OC1 cells. Accumulating evidence indicates that gap junction activity can be altered depending on the amounts of Cx protein, phosphorylation, and internalization (Imanaga et al., 2004). The expression of Cx43 was slightly, but not significantly, decreased in 10 μM cisplatin-treated cells as compared to DMSO-treated control cells (1.6 ± 0.1 vs. 2.0 ± 0.0,
respectively), whereas 25 μM cisplatin significantly decreased Cx43 expression compared to that of DMSO-treated control cells (1.1% ± 0.2% vs. 2.0% ± 0.0%, respectively; Fig. 8A). I next analyzed the localization of Cx43 in cisplatin-treated HEI-OC1 cells. I found that the distribution of Cx43 was changed by exposure to cisplatin in a dose-dependent manner. Cx43 was expressed as scattered spots in the cytoplasm and membrane in DMSO-treated control cells, but was observed to accumulate around the nucleus in cisplatin-treated cells (Fig. 8B). These findings suggested that cisplatin inhibited gap junction communication by inhibiting the trafficking of Cx43 to cell membranes and implied that regulation of gap junctions may affect HEI-OC1 cell survival after cisplatin treatment.
Fig. 7. Change in gap junction communication by cisplatin. (A) Representative
micrographs show prior to bleaching (0 s, left column), immediately after bleaching (25 s, middle column), and after recovery (400 s, right column) in HEI-OC1 cells treated with DMSO, 10 μM cisplatin, or 25 μM cisplatin for 24 h. The region of bleaching is indicated by a dotted line with arrows. Scale bar = 20 μm. (B) Summary of time course of FRAP data for many individual cells. For every bleached cell, the fluorescence signals present in each cell immediately prior to and immediately after photobleaching were normalized to 100% and 0%, respectively. Images are representative of at least 3 separates experiment. (C)
Kymograph analysis of the movement of calcein AM dyes in HEI-OC1 cells treated with DMSO, 10 μM cisplatin, or 25 μM cisplatin for 24 h. Time-lapse images were collected at 10 s intervals before and after photobleaching in the selected ROI. The horizontal lines of the kymograph represent the time of the image series. (D) Lucifer yellow was loaded by scrape line in monolayers of HEI-OC1 cells treated with DMSO, 10 μM cisplatin, or 25 μM cisplatin for 24 h. (E) A bar graph shows the percentage of dye-coupled cells treated with DMSO, 10 μM cisplatin, or 25 μM cisplatin for 24 h. Data are presented as the mean ± SD, P < 0.05; n ≥ 3 independent experiments.
Fig. 8. Cisplatin inhibits the trafficking of Cx43 to cell membranes. (A) Representative
western blotting data for Cx43 protein (upper row) and densitometric analysis (lower row) in HEI-OC1 cells after treatment with DMSO, 10 μM cisplatin, or 25 μM cisplatin for 24 h. β-actin was used as a loading control. Data are representatives of 3 independent experiments and are shown as the mean ± SD, * P < 0.05 versus control. (B) Representative immunofluorescence staining of Cx43 (green) and DAPI (blue) in HEI-OC1 cells after treatment with DMSO, 10 μM cisplatin or 25 μM cisplatin for 24 h.
3. Inhibition of gap junctions reduced cisplatin-induced apoptosis
To examine whether the regulation of functional gap junctions could change cisplatin cytotoxicity in HEI-OC1 cells, I inhibited gap junctions via transfection with Cx43 siRNA and treatment with a gap junction blocker. First, I tested the possible toxic effects of Cx43 siRNA and the gap junction blocker, 18α-GA. Cx43 knockdown or 18α-GA treatment did not affect either cell viability or intrinsic apoptosis-associated molecules, such as cleaved PARP, caspase 3, and p-JNK (Fig. 9). To verify Cx43 siRNA specificity, I tested whether Cx43 siRNA affected the expression level of other cochlear Cx proteins. The protein levels of Cx26 and cx30 were not altered by Cx43 siRNA. I chose 10 μM as the cisplatin concentration yielding an adjustable gap junction, in accordance with Fig. 2 and chose a 48 h cisplatin treatment to evaluate the effects of Cx43 siRNA and 18α-GA in the context of treatment with 10 μM cisplatin. Ie next examined whether knockdown of Cx43 or treatment with 18 α-GA changed cisplatin-induced cell death using an MTS assay. The results revealed that exposure to 10 μM cisplatin significantly decreased cell viability in cisplatin-treated control siRNA-transfected cells (52.2% ± 5.0%) compared to DMSO-treated control cells (100% ± 0.0%), and this decrease was markedly improved by Cx43 knockdown (63.0% ± 3.2%; Fig. 10A). The percent recovery measured by FRAP was decreased in Cx43-knockdown cells, and this decrease was further reduced by 15.1% following exposure to cisplatin (Fig. 10C). Treatment with cisplatin plus 18α-GA (75.0% ± 7.6%) also significantly increased cell viability compared to treatment with cisplatin alone (56.1% ± 2.9%; Fig. 10B). Treatment with 18α-GA plus cisplatin resulted in a significant decrease in FRAP compared to treatment with cisplatin alone (Fig. 10D). This effect was also seen in SLDT assays (data not shown). These findings indicated that gap junctions actively participated in the bystander
effect in response to cisplatin, and this interruption improved cell survival in cisplatin-induced cytotoxicity.
I also found interesting results through quantitative analysis of FRAP data (Fig. 11). First, I analyzed the mobile fraction, defined by the fraction of fluorescent molecules that could undergo exchange between the ROI and the surrounding area, and the velocity of recovery in cisplatin-treated HEI-OC1 cells with or without treatment with gap junction inhibitors using FRAP data. HEI-OC1 cells treated with 10 μM cisplatin showed a positive mobile fraction value and velocity of recovery value, indicating that the function of the cells (i.e., cell-cell communication) could be recovered. On the other hand, HEI-OC1 cells treated with 25 μM cisplatin showed a negative mobile fraction value and velocity of recovery value (Fig. 11A, C). This suggested that cells treated with high concentrations of cisplatin may not be able to recover the ability for cell-cell communication, which may be an important considering factor for selecting the appropriate concentration of cisplatin for cancer therapy. I also observed that treatment with Cx43 siRNA or 18α-GA did not strongly affect the mobile fraction values or the velocity of recovery values (Fig. 11B, D). Namely, HEI-OC1 cells may be able to recover the ability for cell-cell communication even after treatment with gap junction inhibitors, and apoptosis could be prevented. Together, my quantitative analysis of FRAP data suggested that treatment with gap junction inhibitors may represent a new strategy for reduction of cisplatin-induced apoptosis in HEI-OC1 cells.
Fig. 9. Effects of Cx43 knockdown and 18α-GA treatment on cell viability and activation of caspase 3, PARP, and JNK activation. (A) HEI-OC1 cells were transfected
with Cx43 siRNA or control siRNA for 24 or 48 h. Cell viability was quantified by MTS assay. The relative cell viability (%) was calculated according to the viability of control siRNA-transfected cells. The bar graph represents the mean ± SD from 3 independent experiments. (B) The activation of caspase 3, PARP, and JNK was analyzed via western blotting. (C) HEI-OC1 cells were treated with 25 μM 18α-GA or DMSO for 24 or 48 h. Cell viability was quantified by MTS assay. The relative cell viability (%) was calculated according to the viability of DMSO-treated control cells. (D) The activation of caspase 3, PARP, and JNK was analyzed via western blot. Total JNK and β-actin served as controls.
Fig. 10. Effects of blocking gap junction intercellular communication on cisplatin-induced HEI-OC1 cell death. (A) HEI-OC1 cells were transfected with Cx43 siRNA or
h. Cell viability was quantified by MTS assay. The relative cell viability (%) was calculated according to the viability of control siRNA-transfected cells. The bar graph represents the mean ± SD from 3 independent experiments. * P < 0.05 versus control siRNA-transfected cells. (C) Gap junction intercellular communication was measured using FRAP of Cx43 siRNA-transfected or control siRNA-transfected HEI-OC1 cells in the presence or absence of cisplatin (10 μM, 24 h). Scale bar = 20 μm. (E) Summary of time course of FRAP data for many individual HEI-OC1 cells under the conditions above. (B) HEI-OC1 cells were pretreated with 25 μM 18α-GA or DMSO for 2 h and were subsequently treated with or without 10 μM cisplatin for 48 h. Cell viability was quantified by MTS assay. The relative cell viability (%) was calculated according to the viability of DMSO-treated cells. The bar graph represents the mean ± SD from 3 independent experiments. * P < 0.05 versus DMSO control cells. (D) Gap junction intercellular communication was measured using FRAP of HEI-OC1 cells pretreated with 25 μM 18α-GA or DMSO in the presence or absence of cisplatin (10 μM, 24 h). (F) Summary of time course of FRAP data for many individual HEI-OC1 cells under the conditions above.
Fig. 11. Quantitative analysis of photobleaching (FRAP) assay for gap junction activity.
The mobile fraction and velocity of recovery were calculated using FRAP data (Figs. 6C and 6D). Normalized fluorescence recovery for the bleached area (region of interest [ROI]) was calculated with the following formula: mobile fraction = (IE - I0) / (II - I0) (upper), velocity of
recovery = (IT - I0) / time (lower) (Fearon et al., 2011). IE: end value of the recovered
intensity, I0: intensity at time point 0 s, II: initial intensity, and IT: intensity over a time course.
4. Activation of ERK and Akt contributed to inhibition of gap junction-mediated beneficial effects in response to cisplatin
ERK, JNK, and protein kinase B (Akt) are members of the MAPK family and have been reported to be involved in mediating cell survival (Wada and Penninger, 2004). Previous studies have reported that the ERK pathway mediates the bystander effect under toxic conditions (Decrock et al., 2009; Lampe et al., 1991; Wada and Penninger, 2004).
Here, I examined a possible link between the beneficial effects of gap junction blockade and MEK/ERK, PI3K/Akt, and JNK/JUN pathways to identify the protective functions of these pathways. The expression of cleaved PARP was decreased 1.6-fold in Cx43 siRNA-transfected cells compared to control siRNA-transfected cells. Cx43 knockdown also increased ERK (p-ERK) by 1.7-fold and phosphorylated-Akt (p-phosphorylated-Akt) by 1.5-fold compared to transfection with control siRNA. In response to 10 μM cisplatin for 48 h, p-JNK was not changed markedly by knockdown of Cx43 (Fig. 12A). I also examined the effects of 18α-GA on the protein expression of the above targets. Cisplatin treatment of HEI-OC1 cells that has been pretreated with 18α-GA for 2 h resulted in a 1.6-fold decrease in the expression of cleaved PARP compared to that in DMSO-treated control cells. Additionally, the phosphorylation of ERK and Akt was significantly increased by 1.9- and 3.6-fold, respectively, compared to those in DMSO-treated control cells. p-JNK levels were not changed by 18α-GA pretreatment (Fig. 12B). These results suggested that the mechanisms of gap junction inhibition-induced protective effects could be mediated by the activation of ERK or Akt in cisplatin-treated HEI-OC1 cells.
Further supporting these results, I found that the expression levels of ERK and Akt were downregulated by 2 h pretreatment with 20 μM PD98059 (a specific inhibitor of ERK)
and 10 μM LY294002 (a specific inhibitor of PI3K/Akt), respectively. Additionally, pharmacological inhibition of ERK and Akt with PD98059 and LY294002, respectively, abrogated the knockdown of Cx43- or 18α-GA-induced cytoprotection against 10 μM cisplatin for 48 h. As shown above, knockdown of Cx43 with cisplatin caused an increase in cell viability compared to cisplatin alone (73.0% ± 7.8% and 62.3% ± 10.6%, respectively). Inhibition of ERK and Akt reduced the protective effects of knockdown of Cx43 compared to uninhibited cells (43.2% ± 14.3% and 49.0% ± 5.7%, respectively; Fig. 13A). In addition, inhibition of ERK and Akt increased levels of cleaved PARP by 5.3- and 4.0-fold, respectively (Fig. 13C), as compared to uninhibited Cx43-knockdown HEI-OC1 cells treated with cisplatin (Fig. 13C). The protective effects of 18α-GA against cisplatin were also decreased by inhibition of ERK activity compared to uninhibited cells (65.9% ± 9.1% and 36.5% ± 11.0%, respectively; Fig. 13B). Similarly, in 18α-GA-treated HEI-OC1 cells treated with cisplatin, further treatment with PD98059 significantly increased cleaved PARP by 2.1-fold compared to that without inhibition of ERK (Fig. 13D).
Together, these findings demonstrated that the ERK/Akt pathway may play important roles in the cisplatin-induced bystander effect through mediating the activity of gap junctions.
Fig. 12. Effects of blocking gap junction intercellular communication on cisplatin-induced MAPK activation. (A) HEI-OC1 cells were transfected with Cx43 siRNA or
control siRNA for 24 h and were subsequently treated with or without 10 μM cisplatin for 48 h. Western blot analysis was performed using antibodies against PARP, phospho-ERK (p-ERK), total ERK (T-(p-ERK), phospho-JNK (p-JNK), total JNK (T-JNK), phospho-Akt (p-Akt), and β-actin. T-ERK, T-JNK, and β-actin served as controls. On the right side of each panel,
densitometric analysis of the corresponding blot is shown. (B) HEI-OC1 cells were pretreated with 25 μM 18α-GA or DMSO for 2 h and were subsequently treated with or without 10 μM cisplatin for 48 h. Western blot analysis was performed using the same antibodies as above.
Fig. 13. Increased cell viability by knockdown of Cx43 and gap junction blocker treatment was attenuated by ERK and Akt inhibition. HEI-OC1 cells transfected with
Cx43 siRNA (A) and HEI-OC1 cells treated with 18α-GA (B) were pretreated with 20 μM PD98059 or 10 μM LY294002 for 2 h and then exposed to 10 μM cisplatin for 48 h. Cell viability was quantified by MTS assay. The relative cell viability (%) was calculated with untreated control HEI-OC1 cells. * indicates statistically significant differences between 2 groups (P < 0.05). (A and B) Western blot analysis was performed using antibodies against PARP, p-ERK, p-Akt and β-actin. The level of β-actin was used as a loading control.
5. Design of gap junctional and non-junctional condition in auditory cell culture
Cells were seeded at high density (>4 × 104 cells/cm2) and low density (<1 × 104
cells/cm2) to simulate the presence and absence of gap junctions, respectively, because gap
junction channels are formed by cell-to-cell contact. As an initial experiment to optimize these conditions, patterns of Cx43 expression and gap junction communication were tested using two techniques: (a) Fluorescence Recovery After Photobleaching (FRAP), and (b) Scrape Load Dye Transfer (SLDT). As shown in Figure 14A, Cx43 was founded to be widely expressed in HEI-OC1 cells. The gap junction plaques were observed between neighboring cells, while higher levels of Cx43 protein were expressed in high-density cell culture compared with low-density cell culture. Next, I tested the function of gap junctions in the high and low cell density cultures. In the FRAP assay, HEI-OC1 cells recovered about 42% of their pre-bleach signal within 400 s of photobleaching. However, at low cell density, fluorescence intensity did not recover, and was decreased by 16.3% (Fig. 14C). In line with this approach, I performed the SLDT assay at both high and low cell densities. LY dyes are capable of penetrating gap junctions and were added to cell culture media. Dye-containing cells were observed at high cell density, whilst no dyed cells were observed at low cell density (Fig. 14D). Based on this, I next evaluated the effect of junctional and non-junctional Cx43s on cisplatin-induced ototoxicity.
Fig. 14. Junctional and non-junctional Cx43s in cell density-dependent cultures.
HEI-OC1 cells were cultured at high density (>4 × 104 cells/cm2) and low density (<1 × 104
cells/cm2). (A) Western blot analysis of Cx43 expression (upper panel). Equal protein
loading was verified by β-actin expression. Representative confocal immunofluorescent images of Cx43 (green) and DAPI (blue) in the HEI-OC1 cells (lower panel). White arrow shows the intercellular gap junction plaques. The images inside the white square represent a magnified cell. (B) A green fluorescent dyes, calcein AM, was added to HEI-OC1 cells and they were then photobleached with a high-powered focus laser. The region of bleaching is illustrated by dotted lines. A representative confocal image obtained prior to bleaching (pre-bleach, left panel), immediately after bleaching (0 s, middle panel) and after recovery (400 s, right panel) in high cell density (junctional, upper image) and low cell density culture (non-junctional, lower image). The graph represents the percentage of recovery during 400 s (lower panel). (C) LY dye was loaded at the scrape line in high cell density (junctional, left image) and low cell density culture (non-junctional, right image). Histogram shows the
6. Effects of gap junction inhibition on cisplatin toxicity in the junctional and non-junctional condition
To investigate the role of junctional and non-junctional Cx43 on cisplatin toxicity, I used pharmacological inhibition, or interfering RNA during cisplatin treatment. First, I tested the efficiency of Cx43 knockdown by siRNA and inhibition by 18α-GA. The rate of recovery in siRNA-transfected cells decreased by 24.9 % compared to mock-treated cells; however, this was not statistically significant (p > 0.05). Cells treated with 18α-GA also had a recovery rate that was decreased by 64.3 % (p < 0.05). Next, I assessed whether knockdown of Cx43 and treatment with 18α-GA affected the degree of cytotoxicity using the MTS assay. In high cell density cultures, cisplatin-induced cell death was decreased in both conditions following knockdown of Cx43 and treatment with 25 μM 18α-GA (Fig 15A and B). This suggests that gap junctions have a pro-apoptotic role in cisplatin-induced injury in this auditory cell line. My previous studies demonstrated that gap junctions have bystander and protective effects against knockdown of Cx43, and that the effect of 18α-GA against cisplatin is mediated by activation of the mitogen-activated protein kinase (MAPK) pathway. In low cell density cultures, treatment with 18 α-GA had no effect on cisplatin-induced toxicity (Fig. 15C). However, interestingly, the cells in which Cx43 was knocked down displayed higher cell viability compared to mock control cells (Fig. 15D). These findings suggest that Cx43 may play a pro-apoptotic role in the ototoxic effect, which is through a gap junction dependent, as well as independent, mechanisms. To investigate the gap junction independent role of cisplatin-induced ototoxicity, the effects of (a) non-junctional Cx43 hemichannel and (b) non-junctional Cx43 in auditory cell lines during cisplatin-induced toxicity were investigated.
Fig. 15. Effect of 18α-GA and Cx43 knockdown on cisplatin cytotoxicity in the junctional and non-junctional condition. (A and C) HEI-OC1 cells were incubated with 25
μM 18α-GA for 2 h and then co-incubated with 10 μM cisplatin for 48 h. The cell viability was quantified by MTS assay. The relative cell viability (%) was expressed as a percentage relative to the DMSO-treated cells. (B and D) HEI-OC1 cells were transfected with either Cx43 siRNA (si Cx43) or Mock siRNA (si Mock) for 48 h and then incubated with 10 μM cisplatin for 48 h. The relative cell viability (%) was expressed as a percentage relative to the mock-transfected cells. The bar graph shows as mean ± S.D values. * P < 0.05.
7. Cx43 hemichannels was not changed by cisplatin treatment
It has been demonstrated that hemichannels in non-junctional regions can open or close in response to various stimuli, although the physiological role of hemichannels remains to be determined. To investigate the role of hemichannels in pathological perturbations following toxic insults, I used a dye transfer method in HEI-OC1 cells. The functional hemichannels were confirmed by passage of LY dye into cells in the absence of Ca2+ at low
cell density to minimize cell contact. It has been reported that hemichannel activity can be regulated by the extracellular Ca2+ concentration and that opening is induced under low Ca2+
conditions (Valiunas, 2002). Monitoring HEI-OC1 cells showed uptake of LY dyes upon exposure to Ca2+ free media, but not in the presence of normal Ca2+-containing media (Fig.
16A). These LY uptakes were inhibited in cells pretreated with the hemichannel blockers 18α-GA and carbenoxolone (CBX) (Data not shown). These results indicate that Cx43 acts as a hemichannel in a gap junction-independent manner. I then examined whether cisplatin treatment or oxidative stress (via addition of hydrogen peroxide, H2O2) could induce
hemichannel opening. Oxidative stress condition was additionally allows by hydrogen peroxide (H2O2). As shown in Figure 16A, no LY uptake was observed in the presence of
either 100 μM cisplatin or 1mM H2O2. In addition, in cisplatin-treated cells, pre- and
co-treatment with hemichannel inhibitors did not affect cell viability (Fig. 16B). Furthermore, I examined the localization of Cx43 in HEI-OC1 cells using immunocytochemistry. I found that Cx43 was distributed in a scattered pattern in the cytoplasm and plasma membrane, whereas treatment with cisplatin caused significant condensation of Cx43 close to the nucleus. Together, these data suggest that cisplatin inhibits the trafficking of Cx43 to the plasma membrane and that this disturbance influences hemichannel properties. To
investigate the influence of specific Cx43-mediated hemichannels, I transfected Cx-deficient HeLa cells with a Cx43 plasmid. First, I confirmed that the wild type and mock-transfected HeLa cells did not express Cx43, and that the Cx43-transfected HeLa cells expressed Cx43, by both western blot and immunocytochemistry. However, there was no change in the LY uptake during the cisplatin treatment. Furthermore, when cisplatin was administered, pre- or co-treatment with hemichannel inhibitors did not affect the cell viability compared with untreated cells (Fig. 17).
Fig. 16. Change of hemichannel function in HEI-OC1 cells by cisplatin treatment. (A)
LY uptake under normal, no Ca2+, 100 μM cisplatin and 1 mM H
2O2 in HEI-OC1 cells. (B)
MTS assay after exposure to DMSO, 10 μM cisplatin and 10 μM cisplatin with 25 μM 18α-GA for 24 h. Bar graph represents as mean ± S.D values. * P < 0.05.
Fig. 17. Altered hemichannel function following cisplatin treatment in Cx43-transfected HeLa cells. (A) Cx-deficient HeLa cells were transfected with Cx43 plasmid DNA, and
mock plasmid DNA and then incubated for 24 h. Cx43 expression was analyzed by western blot, and normalized to α-tubulin expression (upper panel). Representative confocal immunofluorescent images of Cx43 with DAPI in HeLa cells (lower panel). (B) LY uptake under normal, no Ca2+, 100 μM cisplatin, and 1 mM H
2O2 in Cx43 and mock-transfected
HeLa cells. (C) Live/Dead cell viability assay after exposure to DMSO, 10 μM cisplatin and 10 μM cisplatin with 25 μM CBX for 24 h in Cx43 and mock-transfected HeLa cells. (D) Representative images showing the distribution of live (Calcein AM, green), and dead (Ethidium Homodimer, red) cells. The bar graph represents the percentage of live cells and
dead cells. Data are expressed as mean ± s.d. values. (E) Representative immunofluorescent image of Cx43 and DAPI in Cx43 and mock-transfected HeLa cells after treatment with DMSO and 10 μM cisplatin for 24 h.
8. HeLa cells transfected with cytoplasmic CT domain of Cx43 become sensitive to the cisplatin
The previous experiment was designed to determine the function of Cx43 in the junctional regions compared to that in the non-junctional region. To achieve this, constructs encoding the NT domain of Cx43, comprising amino acids 1–256 and the cytoplasmic CT domain comprising amino acids 243–382 of Cx43 were used (Moorby and Patel, 2001) (Fig. 18A). To determine whether the non-junctional region of Cx43 could be involved in cisplatin-induced toxicity, I expressed the CT domain of Cx43 in Cx-deficient HeLa cells. Immunofluorescence showed that Cx43-CT localized to the cytosol and nucleus, whereas Cx43-NT was mainly localized at the plasma membrane (Fig. 18C). I next examined the effects of these constructs on cisplatin-induced cellular toxicity. As shown in Figure 19A, the cell viability significantly decreased in cisplatin treated mock-transfected cells (91.0 ± 3.3%) compared to DMSO-treated cells (100 ± 2.8%). This decrease was significantly improved by Cx43-FL (78.7 ± 8.5%), NT (85.9 ± 3.9%), and CT (83.3 ± 3.4%) transfection compared to mock transfected HeLa cells (91.0 ± 3.3%) (Fig. 19A). With regard to cell viability, HeLa cells expressing FL, NT, and CT, showed higher levels of cleaved caspase 3 compared to mock-transfected cells (Fig. 19B). These data suggest that both junctional and non-junctional Cx43 may be involved in the cisplatin-induced pro-apoptotic pathway.
Fig. 18. Expression of truncated Cx43 protein in HeLa cells. (A) Schematic
representation of Cx43-FL, NT, and CT. (B) RT-PCR analysis of Cx43-NT (amino acid 1-256, 765 bp) and Cx43-CT (amino acid 243-382, 417 bp) from HeLa cells transfected with mock, Cx43-FL, NT, and CT constructs. (C) Representative confocal immunofluorescent images of Cx43 and DAPI in HeLa cells transfected with mock, Cx43-FL, NT, and CT construct.
Fig. 19. Effect of cisplatin on cell viability in HeLa cells expressing Cx43-FL, NT and CT. (A) HEI-OC1 cells were transfected with mock, Cx43-FL, NT and CT constructs for 24
h and then exposed to 10 μM cisplatin for 24 h. The cell viability was quantified by MTS assay. The relative cell viability (%) was calculated using DMSO-treated cells as a control. (B) Western blot analysis of caspase 3. Equal protein loading was verified by β-actin expression.
9. Cisplatin-induced toxicity decreases following Cx43 knockdown by inhibition of Cx trafficking to the plasma membrane.
Newly synthesized Cx proteins oligomerize into a hexametric structure referred to as a hemichannel at the trans-Golgi network after exiting the endoplasmic reticulum. These hemichannels are then transported along microtubules to the plasma membrane (Fort et al., 2011). BFA is a natural fungal metabolite that blocks the transfer of Cx proteins from the Golgi complex to the plasma membrane, in turn preventing the formation of gap junctions (Smyth et al., 2012). To confirm this in our system, HEI-OC1 cells was incubated with 50 ng /ml of BFA for 24 h, followed which immunostaining was performed against Cx43. As shown in Figure 20A, after treatment of BFA, HEI-OC1 cells no longer had Cx43 at junctional regions and appeared to internalize from the plasma membrane. To quantitatively compare the localization of Cx43 protein in BFA-treated and ethanol-treated HEI-OC1 cells, I drew a line from the center of the nucleus towards the edge of the plasma membrane using the Image J program. Mean fluorescence intensity of Cx43 occurred sporadically in ethanol-treated cells, however, in the presence of BFA, intensities of Cx43 tended to concentrate near the nucleus (Fig. 20B). Under this non-junctional condition, I examined the effect of Cx43 knockdown on cisplatin-induced toxicity. As seen in Fig. 20B, cell viability was increased 1.3-fold in Cx43 knockdown cells compared to mock-transfected cells following exposure to cisplatin at low cell densities. In addition, I observed that cell viability was increased 1.2-fold in Cx43 knockdown cells compared to mock-transfected cells by exposure to cisplatin during co-treatment with BFA (Fig. 20C). Similarly, I showed that cleaved caspase 3 and PARP were decreased by Cx43 knockdown compared to mock-transfection when treated with BFA alone, and in non-treated HEI-OC1 cells (Fig. 20D). These results suggest that
Fig. 20. Effect of Cx43 knockdown on cisplatin-induced cytotoxicity in HEI-OC1 cells in the non-junctional region by BFA. HEI-OC1 cells were treated with 50 ng/mL BFA for
24 h to disrupt to delivery of Cx43 to the cell membrane. (A) Representative confocal immunofluorescent images of Cx43 (green) and DAPI (blue) in BFA-treated cells and ethanol solvent-treated cells. A white square represents a magnified ROI. (B) Analysis of Cx43 fluorescence intensity depending on the distance from the center of the nucleus to the plasma membrane. The dashed line indicates the point away from the nucleus. HEI-OC1 cells were transfected with Cx43 siRNA and mock siRNA for 24 h and then exposed to 25 μM cisplatin with or without pre and co-treatment with BFA. (C) The cell viability was
quantified by MTS assay. Bar graph represents mean ± s.d. values. * P < 0.05. (D) Western blot analysis of PARP, caspase 3, and Cx43. Equal protein loading was verified by β-actin expression. On the right side of western blot images, the densitometric analysis is shown and the bar represents the mean ± S.D. values. * P < 0.05.