Vol. 15, No. 2, November, 2007
□ 증 례 □1)
서 론
멘케스병(Menkes disease)은 꼬인 모발병(kinky hair disease)이라고 불리는, 성염색체 열성유전의 드문 진행성 신경퇴행성 질환으로 운동성 경련, 진 행성 신경학적 악화 양상, 특징적인 모발 이상, 저 체온증, 골격계의 변형, 비정상적으로 꼬인 동맥, 조 기 사망을 특징으로 한다. 이러한 증상은 모두 인체
책임저자 : 권영세, 인하대학교 의과대학 소아과학교실 Tel : 032) 890-3716, Fax : 032)890-2844 E-mail : [email protected]
내 구리 의존 효소들의 결함, 특히 ATP7A 유전자 의 결함과 관련되며 환자의 6-70%에서 대개 3세 이내에 사망한다1). ATP7A 유전자는 Xq13.3 위치 에 있으며 구리 수송 P형 세포막 ATPase(MNK) 를 암호화한다2). 이 ATPase라는 단백질에 결함이 생겨 세포막을 통해 구리를 전달하지 못하므로 많 은 구리 의존 효소들의 기능이 저하된다. 이로 인해 다양한 병리학적 변화가 발생하는데, 동맥은 꼬이고 내강은 불규칙해진다. 이는 중요 효소의 결함으로 탄성 섬유와 교원질의 가교 결합 결여로 발생한다.
뇌 안의 변화는 국소적 회백질 변성이 나타나며 이
영아 연축을 동반한 멘케스병 1례
인하대학교 의과대학 소아과학교실, 울산대학교 의과대학 소아과학교실*
안소현·박신영·강성길·이지은·권영세·손병관·유한욱*
= Abstr act =
A Case of Menkes disease with Infantile Spasm
So Hyun Ahn, M.D., Sin Young Park, M.D., Sung Gil Kang, M.D., Ji Eun Lee, M.D.
Young Se Kwon, M.D., Byung Kwan Son, M.D. and Han Wook Yoo, M.D.*
Department of Pediatrics, College of Medicine, Inha University, Incheon, Korea Department of Pediatrics
*, College of Medicine, Ulsan University, Seoul, Korea
Menkes disease, so called kinky-hair syndrome, is a rare, genetic and progressive neurodegenerative disorder. It is caused by a mutation in the ATP7A gene, which codes for the copper transporting ATPase in the cell organelles. The dysfunction of many cop- per-dependent enzymes results in low concentration of copper in some tissues and accu- mulation of copper in others.
We report a boy presented with kinky hairs, developmental delay, hypotonia and con- nective tissue abnormalities at the age of 4 months. Despite the treatment with various antiepileptic drugs, atonic seizures still persisted. At the age of 7 months, his atonic sei- zures was changed into extensor spasms with modified hypsarrhythmia for some years.
The seizure were controlled by topiramate and vigabatrin. At the age of 22 months, serum copper and ceruloplasmin rechecked as 17 ug/dL(80-150 ug/dL) and 7.3 mg/dL(20-46 mg/
dL) respectively.
The gene study showed ATP7A mutation and the patient was diagnosed as Menkes disease so that copper-histidine was daily injected.
We experienced a case of a 4-month-old boy with Menkes disease and infantile spasm, confirmed by ATP7A mutation.
Key Words : Menkes disease, Infantile spasm, ATP7A mutation
는 뉴런 손실과 교증(gliosis), 그리고 관련된 백질 에서의 축색 변성으로 나타난다3).
증상은 신생아기 특히 생후 첫 수 주경에 나타나 며 운동성 경련 형태로 시작한다. 경련 양상은 간대 근경련성, 때때로 긴장-간대 양상으로 나타난다. 경 련은 멘케스병에서는 흔하나, 경련의 세부 양상에 대한 보고는 거의 없는 실정이다4).
최근 저자들은 ATP7A 유전자 돌연변이로 확진 된, 영아 연축을 동반한 멘케스병 1례를 문헌 고찰 과 함께 보고하는 바이다.
증 례
환 아 : 정○○, 남아, 4개월
주 소 : 3일간의 기면 상태와 수유량 감소 현병력 : 10일 전부터 근력 감소 소견을 보였고, 3일 전부터 시작된 발열과 현저한 수유량 감소 및 기면 상태를 주소로 입원하였다.
과거력 및 가족력 : 재태기간 36주, 출생 체중 2.58 kg으로 제왕절개로 출생하였고, 출생 시 태변 흡입증후군으로 인공호흡기 치료를 4일간 받았다.
산모의 양수 과소증이 있어 시행한 복부 초음파에 서 양측 수신증으로 진단되었다. 양측 부모에게 특 별한 병력은 없었다.
진찰 소견 : 입원 당시 체중 9.1 kg(3백분위수 미 만), 신장 88 cm(90백분위수)이었다. 통통하고 흰
얼굴과 장밋빛 뺨, 꼬이고 푸석거리는 밝은 갈색 모 발을 보였다. 신경학적 진찰 소견에서 의식은 명료 하였고 사지의 근력은 떨어졌으나, 근긴장도와 심부 건 반사는 정상이었다(Fig. 1).
검사 소견 : 말초 혈액 검사에서 백혈구 10,900/
mm3, 혈색소 11.4 g/dL, 적혈구 용적률 34%, 혈소 판 212,000/mm3이었고, 전해질 및 혈청 생화학 검 사는 정상이었고, 혈청 구리 농도 96 ug/dL와 ce- ruloplasmin 농도 4.6 mg/dL로 모두 정상이었다.
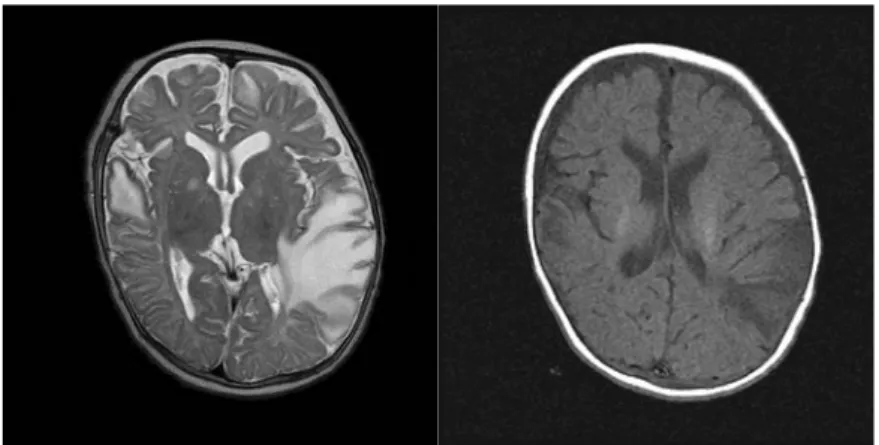
방사선 소견 : 뇌 자기공명영상에서는 양쪽 전두 엽과 측두엽의 부종변화와 왼쪽 측두엽의 국소 뇌 회 조영 증가 소견 그리고 왼쪽 기저핵의 환형 병 변을 보였다(Fig. 2).
Fig. 2A. Brain MRI shows edematous changes in both frontal and temporal lobe.
Fig. 1. The picture shows that his face has a cherubic appearance with a depressed nasal bridge and reduced movement.
뇌파 검사 : 입원 당시 초점성 극서파를 보였다.
치료 및 경과 : 집중치료실 입원 후 이완성 경련 이 동반되어 시행한 뇌척수액 검사는 정상이었으나 열이 지속되어 항생제와 항경련제(phenobarbital) 를 사용하였고, 2병일 시행한 뇌 전산화단층촬영과 뇌 자기공명영상에서 양쪽 전두엽과 측두엽의 부종 변화와 왼쪽 측두엽의 국소 뇌회 조영 증가 소견 그리고 왼쪽 기저핵의 환형 병변이 보였다(Fig. 2A).
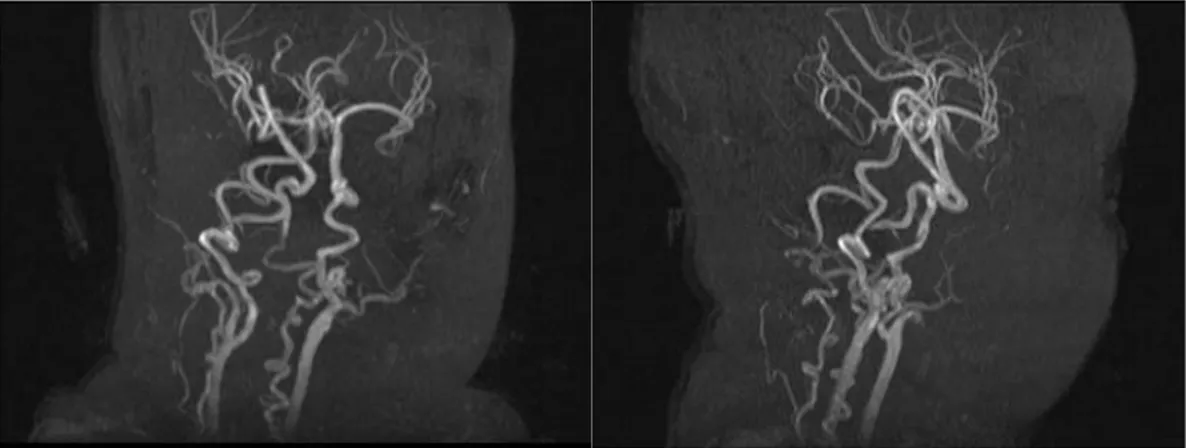
자기공명 혈관촬영술에서 척추 동맥과 대뇌 동맥 그리고 원위분지 혈관의 꼬인 혈관 구조를 보여 멘
케스병을 의심하였으나(Fig. 2B) 혈액 검사에서 정 상 소견을 보이고, 뇌파에서는 비특이적인 초점성 극서파를 보여 뇌염과 대사성 질환 가능성을 고려 하였다. 퇴원 후에도 2차례 구강 칸디다증과 폐렴으 로 입원하였다. 이후 경련이 조절되지 않아 항경련 제를 증량하던 중 생후 7개월경 경련 양상이 신전 양상 연축성 경련으로 변하면서 변형된 고부정 뇌 파 소견을 보였다(Fig. 3). 항경련제를 topiramate 와 vigabatrin로 변경 후 11개월경부터 경련은 조 절되었다.
Fig. 2B. MRA shows tortuous vascular structures of vertebral artery and its distal branch vessels.
Fig. 3. EEG; Hypsarhythmic pattern- multifocal high-amplitude and poly- morphic activity.
22개월경 재시행한 혈청 구리 및 ceruloplasmin 은 각각 17 ug/dL(80-150 ug/dL)과 7.3 mg/dL (20-46 mg/dL)로 감소되었다. 말초혈액을 이용하 여 시행한 ATP7A 유전자 검사(PCR sequencing) 에서 guanine이 adenine으로 바뀐 missense mu- tation(Fig. 4)으로 진단하였다. 출생 시부터 수신증 진단 받았고, 2세경 요로감염으로 입원하여 검사한 배설성 방광요로역류검사에서 다발성 방광게실 소 견 보여 수술하였다. 현재 copper-histidine 피하주 사 및 부적절한 식이로 인한 영양실조로 케모포트 및 경피적 내시경 위루설치술 시행하였고, 골다공증 에 의한 대퇴부 골절에 대한 정형외과적 치료를 하 면서 경과 관찰 중이다.
고 찰
Menkes가 처음 기술한 멘케스병3)은 구리 전달 체(Menkes Cu ATPase)의 결함으로 구리의 결핍 및 구리 함유 효소의 기능 저하로 발생하는 드문 진행성 신경퇴행성 유전 질환이다. 구리 대사 이상 으로 발생하며 이는 ATP7A 유전자의 결함과 관련 있다. 성염색체 열성으로 유전되고, 특히 X염색체 장완(Xq13.2-q13.3)에 위치하는 구리 전달 단백을 암호화하는 유전자의 돌연변이로 발생한다2). ATP7A는 구리 수송 P형 세포막 ATPase(MNK) 를 암호화하며, 뇌를 포함한 대부분의 조직에 분포 하나 간에는 존재하지 않는다. P형 ATPase는 세포 Fig. 4. Partial DNA sequence analysis of ATP7A gene : Genomic DNA
was isolated from the cultured skin fibroblasts of three patients and the peripheral blood of three patients and normal subjects. We performed a gene analysis in 23 exon of ATP7A. After DNA sequencing, a missense mutation as c.4005+5G>A in intron 20 was detected(presented as black circles). Electrophoresis and analysis of the reaction mixtures were done and then patient s gene was larger as 204bp than normal gene(indicated by thick black arrow). After usng RT-PCR product from fibroblast, we found that 204bp in exon 20 was skipped.
막 단백질의 한 구성원으로 세포질과 소포체막을 가로질러 수송하는 역할을 한다. 점막 세포에서 혈 액으로 흡수될 때 Menkes gene product(ATP7A) 가 필요하나 구리 전달체(Menkes Cu ATPase) 결 함으로 세포막을 가로질러 구리를 전달하지 못하므 로, 많은 구리 의존 효소들의 기능이 저하된다. 그 결과 분절된 동맥 내막, 피부와 모발의 색소 결핍, Kinky hair(꼬인 머리카락), 저체온증, 골격의 기형 등이 나타난다.
멘케스병 환자의 6-70%에서 유전자 이상을 발 견할 수 있고, 국내외에서 멘케스 병과 관련한 사례 들이 여러 차례 보고되고 있으나, 유전자검사를 통 해 Guanine이 Adenine으로 바뀐 missense muta- tion으로 확진된 증례는 국내에서 처음 보고하는 것 으로 그 의미가 있다. 멘케스병은 혈중 구리농도의 감소와 특징적인 모발 소견으로 진단이 가능하지만 확진을 위해서는 유전자 검사가 필요하다2, 12).
멘케스병은 대부분 정상적이거나 조산으로 출생 되며, 출생 시 저체온증과 고빌리루빈혈증을 보이 고, 6-12주까지는 정상적으로 성장하고 발달한다.
그러나 생후 2-3개월 사이에 발병하여 대부분 1-3 세 이전에 사망하게 된다5). 환자의 6-70%에서 대 개 3세 이내에 사망한다1). 주로 구리결핍으로 인한 감염 위험의 증가와 불충분한 영양섭취로 사망률이 증가하는 것으로 추정된다. 본 환아의 경우 4개월경 부터 증상이 나타나기 시작하였고 22개월에 진단 받았으며 현재 3년 이상 생존하고 있다. 위식도 역 류 및 반복되는 흡인성 폐렴으로 수차례 입원하였 으나, 케모포트 및 경피적 위루설치술을 시행하여 부적절한 식이로 인한 영양실조를 조기에 해결하여 현재까지 생존하고 있다. 본 증례와 같이 만성 질환 환자의 경우 영양학적인 지지가 생존에 중요한 것 으로 사료된다. 진단은 특징적인 모발과 임상 증상 으로 가능하다. 임상 양상으로 꼬이고, 윤기가 없으 며, 잘 부서지는, 색소가 결핍된 머리카락을 가지며, 특이한 얼굴 모양과 퇴행성 신경장애를 보인다. 발 생빈도는 남아에서 100,000-250,000명에 1명 정도 이다6). 혈중 구리와 ceruloplasmin의 수치가 낮고, 모발을 현미경으로 보면 부서지기 쉽고, 꼬여 있는
양상을 볼 수 있다. 또한 X선 검사를 통해 뼈의 변 형을 확인하고5), 뇌 자기공명영상과 전산화 단층촬 영을 통해 뇌연화증 및 회백질의 퇴행, 뇌의 위축, 뇌막하의 혈종 등을 확인한다7, 8). 5개월 이후부터는 뇌파 검사에서 고부정뇌파(hypsarrhythmia)를 보 였다. 산전 진단은 융모막 검사와 양수 검사를 통해 구리의 수치를 확인할 수 있다9).
본 증례의 경우 처음에 이완성 경련이 동반되었 고, 혈중 구리 농도 및 ceruloplasmin 농도가 정상 이었으나, 생후 7개월 때는 신전 양상 연축성 경련 으로 변하면서 변형된 고부정 뇌파 소견을 보였다.
지속되던 경련은 vigabatrin을 투여 후 조절되었다.
특이할 사항은 처음부터 영아성 연축이 나타나지 않았고 이완성 경련양상이 변하여 고부정 뇌파 소 견을 보이는 경련 형태를 보였다는 것이다. Nadia Bahi 등9)의 연구에 의하면 멘케스병에서의 간질 발 작은 크게 조기, 중기, 말기의 3기로 나누어 볼 수 있고 'two step evolution epilepsy'로 정의된다.
조기 단계(3개월)는 대개 열로 시작되는 국소성 간 대성 간질 중첩증 형태로 나타나며, 발작기간의 뇌 파는 서파형태로 대뇌 후면에서 발생하며, 발작간 뇌파는 다초점성 다형성 서파, 혹은 혼합된 서파 형 태였다. 중기(10개월)에서 경련은 난치성의 영아 연 축형태로 나타나며, 발작간 뇌파는 변형된 고부정 뇌파, 미만성 불규칙한 극서파였고 그 중 6명은 15 개월 이전에 사망하였다 한다. 후기 단계는(25개월) 다초점성 경련, 긴장성 연축, 근간대성 경련 등으로 나타났으며 그 중 2명은 경련 없이 지냈다. 발작간 뇌파는 다초점성 고진폭 활동(multiple high ampli- tude activity), 불규칙 서파와 혼합 형태였으며 대 개 3.6세에 사망하였다 한다. 본 환아의 경우도 전 기에서는 clonazepam, phenytoin 등 모든 항경련 제에 반응이 없고 간기가 되면서 phenobarbital, valproate 모두에 반응이 없었다. 본 증례의 경우 phenobarbital, carbamazepine, topiramate에는 반 응 없었으나 vigabatrin 추가 시 경련이 조절되었 다.
멘케스병에서 간질의 병태생리적 기전은 아직 알 려져 있지 않다10). 그러나 구리결핍과 관련이 있으
며, lysyl oxidase, superoxide dismutase, cyto- chrome coxidase와 관련 있는 것으로 추정된다고 한다1). 특히 lysyl oxidase에 의한 혈관 변화로 혈 전증이 생기고 혈액 공급이 감소된다고 한다. 영아 연축의 경우 많은 사람들이 경련을 일으키는 급성 피질하 손상은 혈관 기능 이상과 에너지 생산 결손 양측에 의해 일어난다고 한다1). 신경 병리학적 변 화는 기저핵과 시상핵에서 축색변성에 의하여 발생 한다. 대뇌 피질의 간근대발작은 진행하는 대뇌피질 변성과 관련 있다고 추정된다.
아직까지 만족할 만한 치료법은 없지만, 가능한 조기에 발견하여 피하로 copper-histidine를 투여하 는 방법이 있다6). 하지만 유전적 결함이 심하거나, 병이 진행된 상태의 환자에게는 유용하지 않으며, 아직까지는 구리요법으로서 병의 진행을 막을 수는 없으나, 생명을 좀 더 연장시켜 줄 수 있다1, 11). 다 른 치료방법으로는 증상적인 치료나 지지적인 치료 를 할 수 있다.
본 증례는 c.4005+5G>A 유전자 돌연변이가 증 명된 첫 멘케스병 환자로 영아 연축으로 항경련제 롤 조절 중이며, 현재 나이는 3년 8개월로 생존하고 있다.
요 약
저자들은 기면과 수유량 감소를 주소로 입원한 4 개월 남자 환아에서 ATP7A 유전자 돌연변이로 확 진된, 영아 연축을 동반한 멘케스병 1례를 경험하였 기에 문헌 고찰과 함께 보고하는 바이다.
References
1) Swaiman KF, Dyken PR. Degenerative diseases primarily of gray matter. In: Swaiman KF, Ashwal S, editors. Pediatric neurology: princi- ples and practice 3rd ed. St. Louis: Mosby 1999:833-5.
2) Watanabe A, Shimizu N. Identification of three novel mutations in Japanese patients with Menkes disease and mutation screening by denaturing high performance liquid chromato- graphy. Pediatr Int 2005;47:1-6.
3) Menkes JH, Wilcox WR. Inherited metabolic disease of the nervous system. In: Menkes JH, Sarnat HB, Maria BL, editors. Child neu- rology. 7th ed. Philadelphia: LWW 2006:115-7.
4) Safaello I, Castelnau P, Blanc N, Ogier H, Evrard P, Arzimanoglou A. Infantile spasms and Menkes disease. Epileptic Disord 2000;2:
227-30.
5) Bacopoulou F, Henderson L, Philip SG. Menkes disease mimicking non-accidental injury. Arch Dis Child 2006;91:919.
6) Sheela SR, Latha M, Liu P, Lem K, Kaler SG.
Copper-replacement treatment for symptomatic Menkes disease: ethical considerations. Clin Genet 2005;68:278-83.
7) Lee ES, Ryoo JW, Choi DS, Cho JM, Kwon SH, Shin HS. Diffusion-weighted MR imaging of unusual white matter lesion in a patient with Menkes disease. Korean J Radiol 2007;8:
82-5.
8) Geller TJ, Pan Y, Martin DS. Early neuro- radiologic evidence of degeneration in Menkes disease. Pediatr Neurol 1997;17:255-8.
9) Bahi-Buisson N, Kaminska A, Nabbout R, Barnerias C, Desguerre I, De Lonlay P et al.
Epilepsy in Menkes disease: analysis of clinical stages. Epilepsia 2006;47:380-6.
10) Schlief ML, West T, Craig AM, Holtzman DM, Gitlin JD. Role of the Menkes copper-trans- porting ATPase in NMDA receptor-mediated neuronal toxicity. Proc Natl Acad Sci 2006;
103:14919-24.
11) Olivares JL, Bueno I, Gallati S, Ramos FJ.
Late-onset treatment in Menkes disease: is there a correlation between genotype and re- sponse to therapy? Clin Genet 2006;69:363-6.
12) You JH, Yoo HW, Kim KS, Kim YW, Kim EY, Paek H et al. 3-year follow-up of a Men- kes disease patient. J Korean Child Neurol Soc 2007;15:94-101.