1)
Introduction
Krabbe disease, also called globoid cell leuko- dystrophy, is a lysosomal storage disorder invol- ving the white matter of the peripheral and cen- tral nervous systems. Krabbe disease is caused by mutations in the galactocerebrosidase gene located on human chromosome 14q31(OMIM
#245200). Typically, the disease occurs during early infancy and progresses rapidly; it invariably has a fatal course. Infants with Krabbe disease are normal at birth. However, symptoms begin between the ages of three and six months with extreme irritability, fever, limb stiffness, seizures, feeding difficulties, vomiting, and slowing of mental and motor development
1). Human galacto-
: 2009 9 4 , : 2009 10 20
: ,
Tel : 02)3410-3539, Fax : 02)3410-0043 E-mail : [email protected]
cerebrosidase complementary DNA was cloned in 1993-1994
2, 3), and many disease-causing mu- tations and functionally silent polymorphisms have been identified
1). In the present report, we describe a case of Krabbe disease with a novel GALC gene mutation.
Case Report
A 6-month-old female infant was admitted to the hospital with inconsolable crying, severe irri- tability, limb stiffness, and gradual loss of de- velopmental milestones. She was born spontaneo- usly from non-consanguineous Korean parents at a gestational age of 39 weeks and with a birth weight of 2,200 grams. The pregnancy was com- plicated by gestational hypertension; however, no abnormal fetal ultrasound findings were reported. There were no perinatal problems. The
A Korean Case of Infantile Krabbe Disease with a Novel GALC Gene Mutation
Soo-Han Choi, M.D., Jeehun Lee, M.D., Sanggoo Lee, M.D.
Chang-Seok Ki, M.D.* and Munhyang Lee, M.D.
Department of Pediatrics and Department of Laboratory Medicine and Genetics*, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Republic of Korea
= Abstract =
Krabbe disease is a rare autosomal recessive neurodegenerative disorder caused by mu- tations in the galactocerebrosidase(GALC) gene. The deficiency of GALC activity leads to the accumulation of psychosine, resulting in apoptosis of myelin-forming cells of the central and peripheral nervous system. The patients with typical infantile onset Krabbe disease have extreme irritability, developmental regression, spasticity, and seizures with an onset prior to six months of age. These children usually die within two years after birth. We report a female infant who showed the characteristic clinical manifestations, disease course, and neuroimaging features of infantile onset Krabbe disease that was confirmed by the identification of a compound heterozygous mutation of the GALC gene.
Key Words : Krabbe disease, Galactocerebrosidase, Mutation
patient was the first baby of this couple and the family history was negative for neurological dis- ease. The baby had normal developmental mile- stones until four months of age: a social smile at two months of age and head control at three months of age. However, there was a gradual increase in irritability, inconsolable crying and limb stiffness starting at four months of age.
Thereafter, the baby began to loose the attained milestones. In addition, the baby had frequent vomiting and episodic febrile events without a definite focus identified.
On physical examination, the baby had poly- dactyly of both hands(six fingers on each hand) but the rest of the physical examination was normal. On admission, the body weight was 6.95 kg(10
th-25
thpercentile), height was 65 cm(25
th- 50
thpercentile) and head circumference was 42 cm(25
th-50
thpercentile). The baby appeared to be alert but did not show any meaningful eye contact or social smile. There were no abnormal findings on the cranial nerve examinations. The motor tone was rigid and spastic in all extremi- ties and axial muscles. The baby had markedly
increased deep tendon reflexes, ankle clonus and a positive Babinski signs.
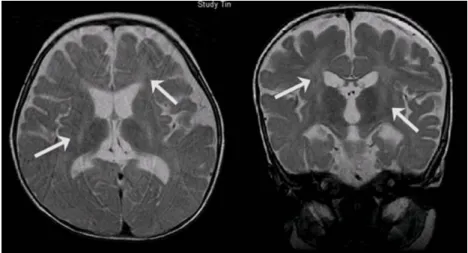
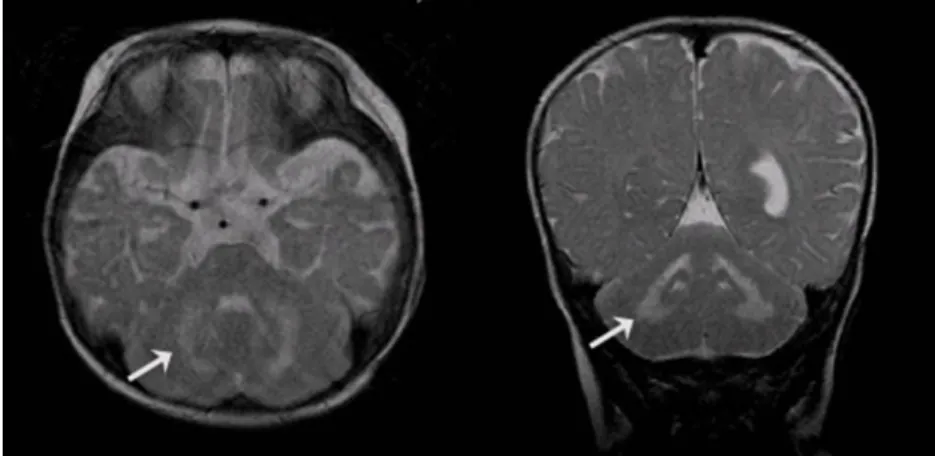
There were no abnormal findings on the blood laboratory test results. The tandem mass spec- trometry screening for inherited metabolic dis- ease was normal. The brain MRI, T2-weighted images showed abnormal high signal intensity of the posterior rims of the internal capsules, bilaterally, the deep white matter(Fig. 1) and the cerebellar white matter(Fig. 2). The electroence- phalography revealed diffuse and asymmetric slow waves in the right and left hemisphere without epileptiform discharges. There were no abnormal findings on the needle electromyo- graphy but the nerve conduction study revealed marked slowing of both the sensory and motor conduction velocities, findings compatible with a diffuse peripheral neuropathy.
A neurodegenerative disease such as Krabbe or other metabolic disorders was suspected based on the history, physical examination, brain MRI, and electrophysiological findings. We performed GALC gene analysis to rule out Krabbe disease;
genomic DNA was extracted from the patients
Fig. 1. The T2 weighted axial and coronal images showed abnormal high signal intensity in the posterior rim of the internal capsule, bilaterally, and in the deep white matter(arrows).
peripheral blood leukocytes. Mutation analysis of the GALC gene on chromosome 14q31 was performed by direct sequencing of all 17 exons.
The analysis of the GALC gene showed two different mutations(Fig. 3): 1) A point mutation T1853C(T>C change at cDNA position 1853) within exon 17, which causes a single amino acid substitution from
618leucine to serine(c.1853T>C;
p.Leu618Ser), and 2) A 9 base pair deletion (TCTGGGAGT) at the cDNA position from 638 to 646 within exon 7, which causes three amino acid deletions from
213leucine to
216serine(c.638_
646del; p.Leu213_Ser216del). This resulted in a
compound heterozygote mutation of the GALC gene. Genetic studies of the parents revealed that these two mutations were inherited from the mother and father. Therefore, Krabbe disease was confirmed by the genetic analysis at 10 months of age.
The baby rapidly regressed physically and de- velopmentally and was bedridden by eight mon- ths of age. In addition, she developed generalized tonic clonic seizures at eight months of age. The electroencephalography showed frequent sharp waves, spikes and polyspike discharges in multi- ple areas. The seizures were controlled with
Fig. 2. The T2 weighted axial and coronal images showed abnormal highsignal intensity in the cerebellar white matter(arrows).
Fig. 3. Direct sequencing of all 17 exons of GALC gene on chromosome 14q31 revealed a c.638_646 del novel mutation in the father(left) and a c.1853T>C known mutation in the mother(right).
phenobarbital. The baby had feeding difficulty and had frequent pulmonary aspiration; she re- ceived conservative management, and occasion- ally was hospitalized for febrile episodes and respiratory problems. The patient died at three years of age after a sudden respiratory arrest.
Discussion
Krabbe disease is one of the classic genetic leukodystrophies first described by Krabbe in 1916
4). It is inherited in an autosomal recessive manner. It occurs in about 1 in 100,000 live births in the United State and a higher prevalence, about 1 in 6,000, has been reported among some Arab communities in Israel
5). In Korea, five cases of Krabbe disease(one case of infantile, three cases of late infantile and one case of juvenile group) have been reported to date
6-10).
The disease results from mutations in the gene that codes for GALC, which results in decreased levels of enzymatic activity. GALC is responsible for the lysosomal hydrolysis of galactosylcera- mide and psychosine. Psychosine accumulation is toxic and causes loss of myelin and infiltration of globoid bodies in the white matter, which re- sults in the signs and symptoms associated with Krabbe disease. The MRI is useful for evaluating the extent of demyelination, and nerve conduction studies show markedly reduced conduction velo- city
1, 11, 12).
The clinical subtypes are divided by onset into the infantile onset type and late onset type. Late onset Krabbe disease includes the late infantile group(onset is between 6 months and 3 years), juvenile group(onset is between 3 and 8 years) and adult group
1). Approximately 85-90% of in- dividuals with Krabbe disease have the infantile form presenting with extreme irritability, spasti-
city, and developmental regression prior to six months of age. The others have symptom onset between the age of six months and the fifth de- cade
5). In our case, the patient showed extreme irritability, limb stiffness and loss of attained milestones by four months of age, which is com- patible with the diagnosis of infantile onset Krabbe disease.
Hagberg et al.
13)divided the clinical course of the infantile form into three stages. Stage I is characterized by hyperirritability, hyperesthesia, episodic fever of unknown origin, and some stiff- ness of the limbs. Rapid and severe motor and mental deterioration develops during stage II.
There is marked hypertonicity and hyperactive tendon reflexes. Tonic or clonic seizures can occur and optic atrophy and sluggish papillary reactions to light are common. In stage III, the patients become decerebrate and blind and have no contact with their surroundings. The average age of death in children with the infantile form is 13 months; patients rarely survive for more than two to three years despite the best suppor- tive care. The disease course of our patient fol- lowed these three stages. For the late-onset forms, individuals can be clinically normal until weakness, vision loss, and intellectual regression become evident. The phenotypes can differ con- siderably between individuals with late-onset forms, even among siblings, who have the same GALC genotype
5).
The diagnosis of Krabbe disease is confirmed by the findings of very low GALC activity(0-5
% of normal mean of 4.2 nmol/h/mg protein) in leukocytes isolated from whole blood and in cul- tured skin fibroblasts or the genetic analysis of the GALC gene
12).
Treatment of individuals with infantile-onset
Krabbe disease that are diagnosed as Stage II or
III is limited to supportive care. Hematopoietic stem cell transplantation(HSCT) in presympto- matic infants and older individuals with mild symptoms has been reported to improve out- comes compared to the more conventional symp- tomatic treatment
14, 15). After HSCT, normal do- nor microglia replaces the affected microglia in the central nervous system and deliver normal enzyme to the surrounding cells; this eventually leads to the discontinuation of demyelination
15). According to previous studies, the transplantation of umbilical-cord blood from unrelated donors to newborns with infantile Krabbe disease favorably altered the natural history of the disease
16, 17). Krabbe disease is inherited in an autosomal recessive manner; therefore genetic counseling and discussion about the availability of prenatal diagnosis are very important in at risk families.
Prenatal diagnosis is possible either by the mea- surement of GALC enzyme activity in a chorionic villi sample, cultured amniocytes or by molecular genetic testing
5, 12). The parents received genetic counseling and were advised about prenatal diag- nosis. The human GALC gene is about 57 kb with 17 exons; it codes for 669 amino acids.
Nearly 70 mutations, including polymorphisms, have been identified
5, 18). Putative disease-causing missense mutations are scattered throughout the exons, excluding exon 12, which is the region of proteolytic cleavage
12). Mutations are more com- mon in the heterozygous state. The 30-kb dele- tion, which always occurs with the 501C>T (R168C) polymorphism, makes up approximately 45% of the mutant alleles in populations with European ancestry
19). One mutation, G>A at position 809 resulting in a G>D substitution at amino acid position 270, always results in the late-onset form of Krabbe disease
19). Xu et al.
20)reported GALC gene mutations in 28 Japanese
patients(new mutations in 17 patients and pre- viously reported mutations in 11 patients), the most common mutation was 12Del3Ins. There has been only one report of a GALC gene muta- tion in Korea to date; Nam et al.
9)reported infan- tile Krabbe disease confirmed by identification of GALC gene mutations, which were 12Del3Ins (c.635_646del12ins3; p.NLWE212_215TP) within exon 7, and C904G(c.904C>G; pPro302Ala) within exon 9. In our case, the T1853C(c.1853T>C;
p.Leu618Ser) was inherited from the mother and was a previously reported GALC gene mutation;
however the GALC gene mutation inherited from the father, c.638_646del; p.Leu213_Ser216del, was a novel mutation.
In summary, we treated a female infant with typical infantile Krabbe disease who showed extreme irritability, inconsolable crying, spastici- ty, and loss of developmental milestones starting at four months of age; thereafter, she rapidly deteriorated and died at three years of age. We confirmed the disease by the molecular genetic testing. And we report the novel mutation of GALC gene in a Korean Krabbe disease patient.
한 글 요 약
새로운 GALC 유전자 변이를 보인 영아형 크라베병 1 례
*
최수한 이지훈 이상구 기창석․ ․ ․ *․이문향
(Krabbe disease) (galactocerebrosidase, GALC)
. GALC
(psychosine)
,
.
6 ,
, , 2
.
, GALC
.
References
1) Suzuki K. Globoid cell leukodystrophy (Krab- be's disease): update. J Child Neurol 2003;18:
595-603.
2) Chen YQ, Rafi MA, de Gala G, Wenger DA.
Cloning and expression of cDNA encoding hu- man galactocerebrosidase, the enzyme deficient in globoid cell leukodystrophy. Hum Mol Ge- net 1993;2:1841-5.
3) Sakai N, Inui K, Fujii N, Fukushima H, Nishi- moto J, Yanagihara I, et al. Krabbe disease:
isolation and characterization of a full-length cDNA for human galactocerebrosidase. Biochem Biophys Res Commun 1994;198:485-91.
4) Krabbe K. A new familial, infantile form of diffuse brain sclerosis. Brain 1916;39:74-114.
5) Krabbe disease: Genetics Home Reference.
United States National Library of Medicine.
Available from: URL://http://ghr.nlm.nih.gov/
condition=krabbedisease
6) Jung SY, Moon HK. A Case of Krabbe Dis- ease. J Korean Child Neurol Soc 2001;9:411-5.
7) Lee C-W. A case of Krabbe disease. Won- kwang medicine 2001;16:107-12.
8) Kim JK, Kim DH, Kang BY, Kwon YS, Hong YJ, Son BK, et al. A Case of Krabbe Disease with Infantile Spasm. J Korean Pediatr Soc 2003;46:95-9.
9) Nam KS, Rhu SH, Sung YH, Oh MS, Jeong HW, Lee BC, et al. A Case of Krabbe Disease Confirmed by Identification of Mutations in the Galactocerbroside beta-galactosidase Gene (GALC). J Korean Neurol Assoc 2004;22:167- 71.
10) Yang HS Lee SH, Kang EK, Park YO. Late- onset Krabbe's Disease (Globoid Cell Leuko- dystrophy): A case report. J Korean Acad Rehabil Med 2005;29:531-6.
11) Husain AM, Altuwaijri M, Aldosari M. Krabbe disease: neurophysiologic studies and MRI cor- relations. Neurology 2004;63:617-20.
12) Wenger DA, Rafi MA, Luzi P, Datto J, Cos- tantino-Ceccarini E. Krabbe disease: genetic aspects and progress toward therapy. Mol Genet Metab 2000;70:1-9.
13) Hagberg B SP, Svennerholm L. Diagnosis of Krabbe's infantile leukodystrophy. J Neurosurg Psychiatry 1963;26:195-8.
14) Cartier N, Aubourg P. Hematopoietic stem cell gene therapy in Hurler syndrome, globoid cell leukodystrophy, metachromatic leukodystrophy and X-adrenoleukodystrophy. Curr Opin Mol Ther 2008;10:471-8.
15) Sakai N. Pathogenesis of leukodystrophy for Krabbe disease: molecular mechanism and cli- nical treatment. Brain Dev 2009;31:485-7.
16) Escolar ML, Poe MD, Martin HR, Kurtzberg J. A staging system for infantile Krabbe dis- ease to predict outcome after unrelated umbi- lical cord blood transplantation. Pediatrics 2006;118:e879-89.
17) Escolar ML, Poe MD, Provenzale JM, Richards KC, Allison J, Wood S, et al. Transplantation of umbilical-cord blood in babies with infantile Krabbe's disease. N Engl J Med 2005;352:
2069-81.
18) Wenger DA SK, Suzuki Y, Suzuki K. The Metabolic and Molecular Bases of Inherited Disease. 8th ed. New York McGraw-Hill, 2001:3669-94
19) Wenger DA, Rafi MA, Luzi P. Molecular genetics of Krabbe disease (globoid cell leuko- dystrophy): diagnostic and clinical implica- tions. Hum Mutat 1997;10:268-79.
20) Xu C, Sakai N, Taniike M, Inui K, Ozono K.
Six novel mutations detected in the GALC gene in 17 Japanese patients with Krabbe disease, and new genotype-phenotype correla- tion. J Hum Genet 2006;51:548-54.