A bacterium strain RR4-35, belonging to the family Rhodobac- teraceae was isolated from a biofilter of seawater recirculating aquaculture system (RAS) located in Busan, South Korea. The strain showed low 16S rRNA similarity (< 96.47%) against species with valid names in the family. PacBio RS II sequencing yielded one complete chromosome (3,833,345 bp with 59.3% G + C content) and six plasmids. A total of 4,487 genes, 4,436 CDSs, 47 tRNAs and 3 rRNAs were annotated. Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis detected gene clusters related to pathways such as denitrification and benzoate degra- dation. Genomic analysis might show the potential role of strain RR4-35 in the nitrogen cycle and biodegradation of organic compounds in the RAS.
Keywords: Rhodobacteraceae, RR4-35, complete genome sequence, recirculating aquaculture system
The family Rhodobacteraceae is one of the major bacterial groups in the class Alphaproteobacteria (Garrity et al., 2005).
Rhodobacteraceae was first established by Garrity et al. (2005).
At the time of writing, the family contained 182 genera which have a validly published and correct name including the type genus, Rhodobacter (https://lpsn.dsmz.de/family/rhodobacteraceae).
The family comprises photoheterotrophic and chemoorganotrophic
bacteria in an aerobic environment and anaerobic non-sulfur bacteria involving degradation and mediation of various com- pounds in marine environments (Pujalte et al., 2014). Members of this family were found in diverse environments, especially in marine environments such as seawater (Wu et al., 2015), aquaculture farm (Rhee et al., 2018), brine-sea water interface (Zhang et al., 2017), and deep-sea vent (Takai et al., 2009).
A recirculating aquaculture system (RAS) was a closed aquaculture system that reused the rearing water by purifying water in the system. In the RAS, biodegradation and detoxifi- cation of waste organic matters, nitrogen compounds and feed debris are mainly carried out by bacterial activity (Zhang et al., 2011). A culture-independent study showed that Rhodobacteraceae was one of the major groups in the RAS (Lee et al., 2016).
Rhodobacteraceae was also found to be one of the most predominant bacterial groups (30.2~49.6%) at family level in the bioreactor of RAS for shrimp aquaculture (Chen et al., 2019).
In this study, we obtained and analyzed the whole genome sequences of the strain RR4-35 that was isolated from a biofilter of a seawater RAS and was distantly related to known species.
The analysis of genome will provide a basis for understanding the role of the Rhdobacteraceae bacterium in the RAS.
Strain RR4-35 was isolated from a biofilm sample obtained from the surface of a RAS-biofilter in Busan, South Korea. The
Korean Journal of Microbiology (2021) Vol. 57, No. 1, pp. 58-61 pISSN 0440-2413
DOI https://doi.org/10.7845/kjm.2021.0133 eISSN 2383-9902
Copyright ⓒ 2021, The Microbiological Society of Korea
Whole genome of strain RR4-35, a novel bacterium in the family Rhodobacteraceae
Hyun-Kyoung Jung
1,2†, Young-Sam Kim
1,2†, and Kyoung-Ho Kim
1,2*
1
Department of Microbiology, Pukyong National University, Busan 48513, Republic of Korea
2
School of Marine and Fisheries Life Science, Pukyong National University, Busan 48513, Republic of Korea
Rhodobacteraceae 과의 신균인 RR4-35의 전장 유전체 분석
정현경
1,2†・ 김영삼
1,2†・ 김경호
1,2*
1
부경대학교 미생물학과,
2부경대학교 해양수산생명과학부
(Received December 31, 2020; Revised January 25, 2021; Accepted January 28, 2021)
†
These authors contributed equally to this work.
*For correspondence. E-mail: [email protected];
Tel.: +82-51-629-5611; Fax: +82-51-629-5619
Complete genome of Rhodobacteraceae strain RR4-35 ∙ 59
Korean Journal of Microbiology, Vol. 57, No. 1 sample was serially diluted using phosphate buffered saline
solution (1X PBS; pH 7.4) and incubated on Marine Agar 2216 (MA, Difco) at 28°C for a week. Pure culture was obtained and phylogenetic analysis was conducted as described previously (Kim et al., 2019). The 16S rRNA gene sequence showed low similarities against the species with valid names in Ezbiocloud server (Yoon et al., 2017) such as Roseovarius litorisediminis CECT 8287
T(96.47%), Roseovarius aestuarii CECT 7745
T(96.18%), Roseovarius lutimaris DSM 28463
T(95.62%), and Phaeobacter inhibens DSM 16374
T(95.59%) that all belong to the family Rhodobacteraceae.
Pure isolate cultured on MA at 28°C for 3 days was used to extract genomic DNA using Genomic DNA Prep Kit (BIOFACT Corp.). The genomic DNA was then subjected to library construction and single-molecule real-time (SMRT) sequencing (Macrogen) (Ardui et al., 2018) using PacBio RS II system (Pacific Biosciences). A total of 152,071 subreads (1,358,239,045 total bases; N50, 12,284 bp; mean length, 8,931 bp) were generated using SMRT sequencing and subreads were de novo assembled by Hierarchical Genome Assembly Process (HGAP, Version 3.0, Pacific Biosciences). A total of seven contigs (total base, 4,557,782 bp) consisting of one chromosome (3,833,345 bp with 59.3 mol% G + C content) and six plasmids (168,707 bp, 59.31 mol%; 140,737 bp, 59.5 mol%;
136,456 bp, 59.6 mol%; 126,025 bp, 60.8 mol%; 90,429 bp, 57.0 mol%; 62,083 bp, 62.9 mol%) were obtained after assembly.
All contigs are circular except for contig 6 (Table 1). The genomes were annotated by NCBI Prokaryotic Genome Annotation Pipeline (PGAP; Jan. 2020) (Tatusova et al., 2016)
and a total of 4,487 genes, 4,436 CDSs, 47 tRNAs and 3 rRNAs were annotated from chromosome and plasmids (Table 1).
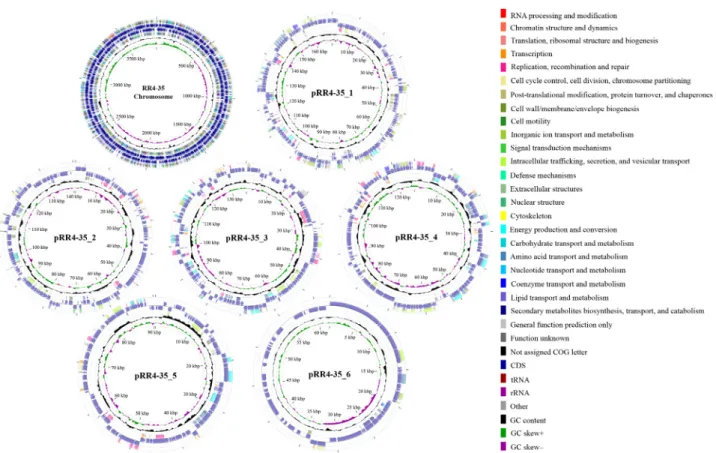
Constructing graphical circular genome maps was performed using CGView Comparison Tool (Grant and Stothard, 2008) (Fig. 1). One CRISPR locus and two CRISPRs candidates were detected by CRISPRFinder (http://crispr.i2bc.paris-saclay.fr) (Grissa et al., 2007). It was detected that one incomplete prophage sequence coding viral components such as tail, head and phage-like proteins using Phage Search Tool Enhanced Release (PHASTER; http://phaster.ca/) (Arndt et al., 2016).
All softwares were used as default parameters unless otherwise indicated.
It was detected that genes for denitrification such as three subunits of respiratory nitrate reductase (narI; WP_165197213.1, narH; WP_165197215.1, and narG; WP_165197216.1), nitrate/
nitrite transporter (narK; WP_165197217.1), nitrite reductase (nirK; WP_165193008.1), two subunits of nitric oxide reductase (norB; WP_165192992.1 and norC; WP_165192994.1) and nitric oxide reductase activation protein (norD; WP_019298187.1) from Kyoto Encyclopedia of Genes and Genomes (KEGG;
https://www.genome.jp/kegg/) (Kanehisa and Goto, 2000) analysis, while nitrous-oxide reductase (nosZ), the enzyme for final step of denitrification that reduces nitrous oxide and produces nitrogen gas, was not observed.
Annotation showed genes related to the benzoate degradation pathway such as genes for 4-hydroxybenzoate 3-monooxygenase (pobA; WP_165192748.1), two subunits protocatechuate 3, 4-dioxygenase (pcaH; WP_165194558.1 and pcaG; WP_
165194560.1), 3-carboxy-cis,cis-muconate cycloisomerase (pcaB;
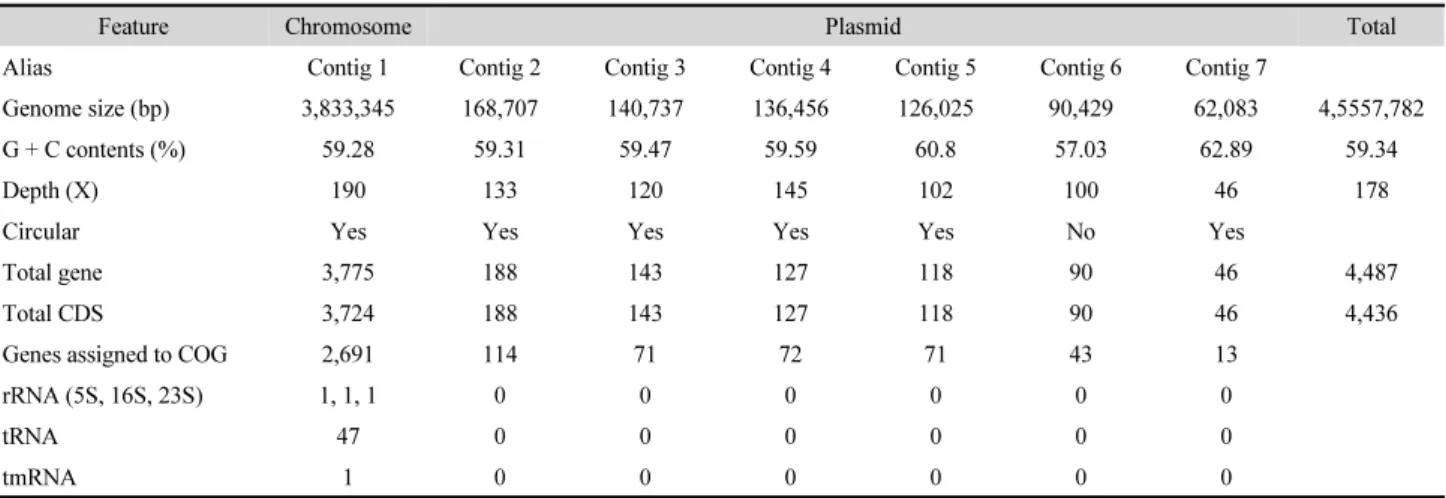
Table 1. Genomic features of strain RR4-35
Feature Chromosome Plasmid Total
Alias Contig 1 Contig 2 Contig 3 Contig 4 Contig 5 Contig 6 Contig 7
Genome size (bp) 3,833,345 168,707 140,737 136,456 126,025 90,429 62,083 4,5557,782
G + C contents (%) 59.28 59.31 59.47 59.59 60.8 57.03 62.89 59.34
Depth (X) 190 133 120 145 102 100 46 178
Circular Yes Yes Yes Yes Yes No Yes
Total gene 3,775 188 143 127 118 90 46 4,487
Total CDS 3,724 188 143 127 118 90 46 4,436
Genes assigned to COG 2,691 114 71 72 71 43 13
rRNA (5S, 16S, 23S) 1, 1, 1 0 0 0 0 0 0
tRNA 47 0 0 0 0 0 0
tmRNA 1 0 0 0 0 0 0
60 ∙ Jung et al.
미생물학회지 제57권 제1호
WP_165194563.1), 4-carboxymuconolactone decarboxylase (pcaC; WP_165194557.1) and 3-oxoadipate enol-lactonase (pcaD; WP_165192750.1), in which we identified the presence of genes responsible for a degradation pathway from 3,4- dihydroxy-benzoate to 3-oxoadipate.
Benzoate biodegradation and nitrate reduction pathways were also detected in Phaeobacter gallaeciensis DSM 17395
T(pcaG; WP_014878970.1, pcaH; WP_014878971.1, HpaR; WP_
014881250.1, nirK; WP_014881756.1, norB; WP_014881752.1 and norC; WP_014881753.1) and some Roseovarius species, included in Roseovarius mucosus SMR3
T(npcC; WP_008281990.1, nasA; WP_081507525.1, nasD; WP_081507524.1 and nosZ;
WP_081508048.1) and Roseovarius indicus DSM 26383
T(pcaH;
WP_057822108.1, pcaG; WP_151175233.1, WP_151175215.1, nirK; WP_057816130.1, norB; WP_057816123.1 and norC;
WP_057816125.1).
Degradation of organic compounds might be coupled with denitrification. A study reported that benzene degradation was
observed in enrichment cultures under nitrate reduction condition (Burland and Edwards, 1999). The whole genome analysis of strain RR4-35 showed its potential role associated to nitrogen cycle and organic compound biodegradation.
Nucleotide sequence and strain accession numbers The strain is available at the Korean Collection for Type Cultures (accession number KCTC 72134). The complete genome sequences including chromosome and plasmids of strain RR4-35 were deposited in DDBJ/EMBL/NCBI GenBank under accession numbers CP049037 (chromosome) and CP049038- CP0490343 (plasmids).
적 요
Rhodobacteraceae 과에 속하는 신종 균주 RR4-35는 해수 순환 여과 양식 시스템(RAS)의 바이오 필터에서 분리되었다.
Fig. 1. Circular map of strain RR4-35 contigs. From the center to the outside: genome label, GC skew (green and purple), G + C content (black), CDSs colored
by COG categories on the reverse strand, CDSs including RNAs on the reverse strand, CDSs including RNAs on the forward strand, CDSs colored by COG
categories on the forward strand.
Complete genome of Rhodobacteraceae strain RR4-35 ∙ 61
Korean Journal of Microbiology, Vol. 57, No. 1 이 균주는 그 과에 속한 유효명을 가진 종들과 낮은 16S rRNA
유전자 유사도를 보여주었다(< 96.47%). PacBio RS II 분석을 통해 하나의 염색체(3,833,345 bp 크기의 G + C 함량 59.3 mol%)와 여섯 개의 플라스미드 서열이 확인되었다. 이 균주 의 유전체들은 총 4,487개의 유전자, 4,436개의 CDS, 47개의 tRNA와 3개의 rRNA 유전자를 포함한다. Kyoto Encyclopedia of Genes and Genomes 분석을 통해 탈질 과정과 벤조에이트 분해와 관련된 유전자 클러스터들이 확인되었다. 본 유전체 분석 결과는 RAS에서 질소 순환과 유기 물질의 분해에 대한 RR4-35 균주의 잠재적인 역할을 보여준다.
Acknowledgments
This work was supported by a Research Grant of Pukyong National University (2019).
References