A novel algicidal bacterium, designated strain BG1, belonging to the family Flavobacteriaceae was isolated from coastal seawater, East Sea of Korea. Its complete genome sequence was determined using PacBio RS II system and Illumina Hiseq X-ten platform. The genome of strain BG1 comprised of a single circular 3,708,790 bp chromosome with G + C content of 37.3%, and encoded 3,160 protein-coding genes, 9 rRNA genes, 44 tRNA genes, 4 non-coding RNA genes and 27 pseudo genes.
The strain BG1 contained various biopolymer degradation-related genes and secondary metabolites biosynthesis-related genes such as terpenoids and polyketide. This genomic information is expected to be useful for application to biopolymers degradation and the screening of bioactive substances.
Keywords: Flavobacteriaceae sp. BG1, complete genome sequence, Illumina HiSeq X-ten, PacBio RS II, secondary metabolites
Red tides are natural global phenomena that have been caused by enormous growth of plankton algae, such as dinoflagellates and diatoms, in marine environments (Anderson et al., 2002).
Heretofore, many algicidal bacteria to be used as biological
control agents have been isolated from various marine environ- ment for decrease and elimination of red tide (Mayali and Azam, 2004; Meyer et al., 2017). Biologically active ingredients isolated from these bacteria have been known as alkaloids, amino acid derivatives, peptides, proteins, enzymes, polyketides, terpenes, and fatty acids (Meyer et al., 2017). In this report, we describe the complete genome sequence and annotation of a novel algicidal bacterium Flavobacteriaceae sp. BG1.
An algicidal marine bacterium against marine diatom Skeletonema costatum, designated strain BG1, was isolated from coastal seawater, East Sea (38° 18’ 41” N, 128° 32’ 33” E) of Korea. The strain BG1 was routinely cultured on marine agar 2216 (Difco) at 30°C for 3 days. Phylogenetic analyses based on 16S rRNA gene sequence indicated that strain BG1 was affiliated to the family Flavobacteriaceae and showed highest similarity to Galbibacter mesophilus Mok-17
T(95.5%).
For the sequencing of the whole genome, the cells were incubated at 30°C in marine broth 2216 (Difco) for 2 days and the genomic DNA was extracted using MagAttract
®HMW DNA kit (Qiagen) according to the manufacturer’s instructions.
The sequencing of the whole genome was performed on the PacBio RS II system and Illumina Hiseq X-ten platform by Macrogen Inc.. The sequencing reads of PacBio were assembled
Korean Journal of Microbiology (2020) Vol. 56, No. 4, pp. 419-421 pISSN 0440-2413
DOI https://doi.org/10.7845/kjm.2020.0122 eISSN 2383-9902
Copyright ⓒ 2020, The Microbiological Society of Korea
Complete genome sequence of a novel algicidal bacterial strain BG1 belonging to the family Flavobacteriaceae
Ji-Sung Oh and Dong-Hyun Roh*
Department of Biological Sciences and Biotechnology, Chungbuk National University, Cheongju 28644, Republic of Korea
Flavobacteriaceae 과에 속하는 신종 살조성 세균 BG1 균주의 유전체 염기서열 완전 해독
오지성 ・ 노동현*
충북대학교 대학원 생명시스템학과
(Received November 25, 2020; Revised December 3, 2020; Accepted December 3, 2020)
*For correspondence. E-mail: [email protected];
Tel.: +82-43-261-3368; Fax: +82-43-264-9600
420
∙ Oh and Roh미생물학회지 제56권 제4호
using the RS Hierarchical Genome Assembly Process 3.0 (HGAP) (Chin et al., 2013), and the sequencing reads of Illumina were applied for accurate genome sequence using Pilon software (version 1.21) (Walker et al., 2014). Genome completeness and contamination were verified with CheckM (Version 1.0.18) (Parks et al., 2015). The genome annotation was performed using NCBI Prokaryotic Genome Annotation Pipeline (PGAP) (Tatusova et al., 2016). Additional function of the predicted genes were conducted by EggNOG 5.0 (Huerta-Cepas et al., 2019), BlastKOALA with KEGG database (Kanehisa et al., 2016) and Rapid Annotations using Subsystems Technology (RAST) server with SEED (Aziz et al., 2008).
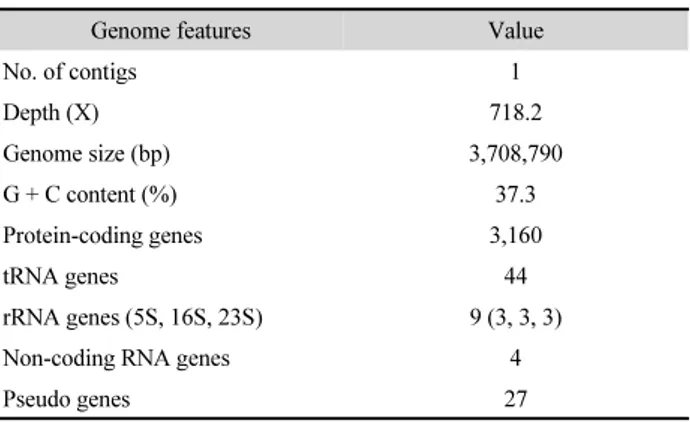
The complete genome of strain BG1 was composed of one contig, 3,708,790 bp in length. The G + C content of the genome was 37.3%. The results of CheckM estimation indicated that genome completeness was 99.67% with 0.17% contamination and 0% strain heterogeneity. A total of 3,160 protein-coding genes, 9 rRNA genes, 44 tRNA genes, 4 non-coding RNA genes and 27 pseudo genes were predicted (Table 1).
The genome sequence of strain BG1 contained carotenoid biosynthesis-related genes such as 15-cis-phytoene synthase CrtB, pytoene desaturase CrtI and β-carotene 3-hydroxylase CrtZ. It contained gliding motility-related genes; gliding motility- associated ABC transporter ATP-binding subunit GldA, gliding motility protein GldB/GldC/GldD/GldE/GldG/GldJ/GldL/GldM/
GldN, gliding motility associated ABC transporter permease protein GldF, gliding motility associated lipoprotein GldH/
GldK, peptidyl-prolyl cis/trans isomerase GldI. It contained also bacterial motility-related genes such as CheA/CheB/CheR/
CheY (involved in chemotaix) and CpaA/CpaB/CpaC/CpaD/
CpaE/CpaF/PilA/PilD (involved in pilus system). Its genome
encoded bacterial secretion system-related genes such as GspA/GspC/GspD/GspE/GspF/GspG/GspH/GspI/GspJ/Gsp K/GspL (involved in Type II secretion system), SecA/SecB/
SecD/SecE/SecF/SecG/SecY (involved in Sec system), and type IX secretion system membrane protein PorP/SprF (in- volved in PorSS, Type IX secretion system). It contained various biopolymer degradation-related genes; α-amylase AmyA/AmyS, cellulase CelC, β-glucosidase BglX, xylose isomerase XylA, xylose kinase XylB, N-acetylglucosamine-6-phosphatase dea- cetylase NagA, glucosamine-6-phosphate deaminase NagB, lipolytic enzyme LipA/LipB. It also encoded biosynthesis of terpenoids and polyketides-related genes; acetyl-CoA-C-acetyl- transferase ACAT, 1-deoxy-D-xylulose-5-phosphate reducto- isomerase Dxr, 4-diphosphocytidyl-2-C-methyl-D-erythritol- kenase IspE, 1-deoxy-D-xylulose-5-phosphate synthase Dxs, (E)-4-hydroxy-3-methylbut-2-enyl-diphosphate synthase IspG, 4-hydroxy-3-methylbut-2-en-1-yl diphosphate reductase IspH, 2-C-methyl-D-erythritol 4-phosphate cytidylyltransferase IspDF, farnesyl diphosphate synthase IspA, dTDP-4-dehydrorham- nose reductase RmlD, glucose-1-phosphate thymidylyltrans- ferase RmlA, dTDP-glucose 4,6-dehydratase RmlB, dTDP-4- dehydrorhamnose 3,5-epimerase RmlC. The genome also contained biosynthesis of secondary metabolites-related genes;
phenylalanine ammonia-lyase PAL, acyl carrier protein AcpP, tryptophan 7-halogenase PrnA, ornithine racemase Orr. These abundant biopolymer degradation genes, protein secretion genes, and secondary metabolite production genes are thought to be useful in biological industry applications.
Nucleotide sequence accession numbers
The strain BG1 is available at KACC 16660, KCCM 43341 and JCM 33635. The whole genome sequence is accessible in GenBank under the accession number CP058364.
적 요
Flavobacteriaceae 과에 속하는 신종 살조성 세균 BG1 균 주는 동해 연안 해수로부터 분리되었고, PacBio RS II system 과 Illumina Hiseq X-ten platform을 사용하여 전체 유전체 염 기 서열을 결정하였다. BG1 균주의 유전체는 37.3% G + C 함 량을 지닌 3,708,790 bp 길이의 단일 유전체로 구성되었고, 전
Table 1. Genome features of strain BG1
Genome features Value
No. of contigs 1
Depth (X) 718.2
Genome size (bp) 3,708,790
G + C content (%) 37.3
Protein-coding genes 3,160
tRNA genes 44
rRNA genes (5S, 16S, 23S) 9 (3, 3, 3)
Non-coding RNA genes 4
Pseudo genes 27
Complete genome sequence of Flavobacteriaceae sp. BG1∙
421
Korean Journal of Microbiology, Vol. 56, No. 4 체 3,160개의 단백질 암호 유전자, 9개의 rRNA 유전자, 44개
의 tRNA 유전자, 4개의 non-coding RNA 유전자 및 27개의 위 유전자(pseudo gene)가 확인되었다. BG1 균주는 다양한 생물 고분자 물질 분해 관련 유전자와 terpenoids, polyketides 및 2 차 대사산물 생합성 관련 유전자가 포함되어 있었다. 이러한 게놈 정보는 생체 고분자 분해 및 생체 활성 물질 스크리닝에 적용하는데 유용할 것으로 예상된다.
Acknowledgments
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2017R1D1 A3B04033871).
References
Anderson DM, Glibert PM, and Burkholder JM. 2002. Harmful algal blooms and eutrophication: nutrient sources, composition, and consequences. Estuaries 25, 704–726.
Aziz RK, Bartels D, Best AA, DeJongh M, Disz T , Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M, et al. 2008. The RAST Server: rapid annotations using subsystems technology.
BMC Genomics 9, 75.
Chin CS, Alexander DH, Marks P, Klammer AA, Drake J, Heiner C, Clum A, Copeland A, Huddleston J, Eichler EE, et al. 2013.
Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 10, 563–569.
Huerta-Cepas J, Szklarczyk D, Heller D, Hernández-Plaza A, Forslund SK, Cook H, Mende DR, Letunic I, Rattei T, Jensen LJ, et al.
2019. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 47, D309–D314.
Kanehisa M, Sato Y, and Morishima K. 2016. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 428, 726–731.
Mayali M and Azam F. 2004. Algicidal bacteria in the sea and their impact on algal blooms. J. Eukaryot. Microbiol. 51, 139–144.
Meyer N, Bigalke A, Kaulfuß A, and Pohnert G. 2017. Strategies and ecological roles of algicidal bacteria. FEMS Microbiol. Rev. 41, 880–899.
Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, and Tyson GW.
2015. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055.
Tatusova T, DiCuccio M, Badretdin A, Chetvernin V, Nawrocki EP, Zaslavsky L, Lomsadze A, Pruitt KD, Borodovsky M, and Ostell J. 2016. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 44, 6614–6624.
Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, Cuomo CA, Zeng Q, Wortman J, Young SK, et al. 2014. Pilon:
an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9, e112963.