P1-26 / J.-S. Kim
IMID 2009 DIGEST •
Abstract
We present a Brownian molecular dynamics computer simulation method for calculating the rotational viscosity of the liquid crystal mixture comprising pentylcyanobiphenol (5CB) and decylcyanobiphenol (10CB). Mean director of the ensemble has been used as a nematic director. Results show a good agreement with experimental ones [Sudeshna DasGupta et al., Physics Letters A 306(2003)235-242].
1. Introduction
The calculation of the rotational viscosity of liquid crystals (LCs) is one of the most challenging works in a MD computer simulation [1-7]. There have been several studies on a single kind of liquid crystals. Methods of calculation of the rotational viscosity for a single kind of liquid crystal, viz., a single element have been studied by Satoru Kuwajima, Atsutaka Manabe (2000)[6], D. L. Cheung, S.J. Clark, M.R.Wilson (2002)[7] and M. Ilk Capar, E. Cebe (2005)[9]. However, there has been a great demand to estimate the rotational viscosity of a mixture comprising two or more different kinds of liquid crystals. This is due to the reason that today’s most of LCD monitors or TVs contain a mixture of 15 to 20 different kinds of liquid crystals for the improvement of their performances such as shorter response time and a broader nematic phase range.
Nematic LC is a rod-like molecule and its local alignment of the long axis along a particular direction is called director.
Fig. 1. Structure of nematic LC 5CB and 10CB.
Fig. 2. A schematic diagram showing the mean director of the ensemble of the nematic liquid crystal mixtures (red: 5CB, blue: 10CB).
2. Computational details
Geometry optimization of the each LC molecule has been performed at the level of hf/6-31G*//hf/6-31G* using Gaussian 03W package. Atomic charges have been determined using RESP scheme and Gaussian input parameters for MD computer simulation were as follows: ‘# opt hf/6-31g (d) scf=tight test pop=Mk iop (6/33=2, 6/42=6)’. We have
A Molecular Dynamics Computer Simulation Method
for the Calculation of Rotational Viscosity
of Liquid Crystal Mixture
Jin-Soo Kim
1, Farzana Ahmad
1, Jamil Muhammad
1, Sang-Woo Park
1,
Jin-Woo Lee
1, Hee-Young Yun
1, Jae Eun Jung
2, Jae Eun Jang
2,
Young-Jae Jeon
1and Yong-Bae Kim
11
Dept. of Chemistry, College of Science, Konkuk University, Seoul, 143-701, Korea TEL:82-2-450-3379, e-mail: [email protected]
2
Samsung Advanced Institute of Technology, Yongin, 449-712, Korea
P1-26 / J.-S. Kim
• IMID 2009 DIGEST
performed MD computer simulation with potential energy equation by using AMBER MD package for Linux as follows:
(
)
(
)
(
)
(
)
∑
∑
∑
∑
∑
< < + ⎟ ⎟ ⎠ ⎞ ⎜ ⎜ ⎝ ⎛ − + + + − + − = atoms j i ij j i atoms j i 6ij ij 12 ij ij dihedrals n 2 eq angles θ 2 eq bonds r , R q q R B R A -n cos 1 2 V θ θ K r r K Uε
γ
φ
(1)where Rij is the distance between the i-th and j-th
atoms of the pair of different molecules and qn is the
electric charge of n-th site [10]. Generalized AMBER force field (GAFF) is different from previous AMBER force field developed for the bio molecules such as protein and DNA. GAFF has been developed for the general organic molecules and it is appropriate for liquid crystal molecules [10]. We have performed Langevin dynamics method to simulate the frictional behaviour of the liquid crystals within the mixture system. Accordingly, MD simulation using the mean director method contains the meaning of the Brownian behaviour of the mean director with frictional effect within the mixture system. Whole processes of the MD computer simulation are as follows:
TABLE 1. MD workflow
Steps Ensemble Time(ns) Purpose 1 Canonical 0.1 To equilibrate the temperature 2 Isothermal-isobaric 2 To raise the density 3 Isothermal-isobaric 2 To equilibrate the system 4 Isothermal-isobaric 2 Brownian dynamics
3. Results and discussion
Figure 3 shows the first step of MD start-up from the artificial structure containing about 350 molecules to equilibrate the temperature of the canonical ensemble system. Second step of MD is to raise the
density of the isothermal-isobaric ensemble system that starts to lose their initial artificial structure which approaches to the real system. The third step of MD is for the equilibrium of the isothermal-isobaric ensemble system.
(a) (b) (c)
Fig. 3. Snapshot of a NLC mixture of (a) initial structure (b) second step of MD (c) fourth step of MD.
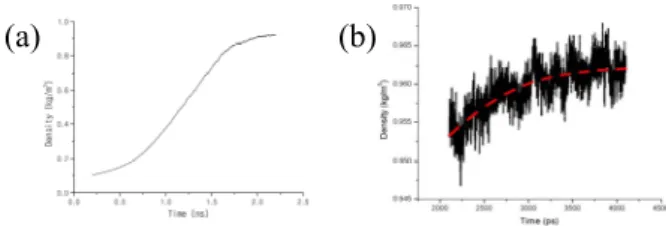
Figure 4 shows that the density dramatically rise during the second step of MD (a) and the third step of MD (b). Equilibrium process of the third MD is one of the most important step in the present work. Density must converge stably. The second and the third step of MD are the same NPT processes, but we have divided it into two steps to investigate the each fluctuation of the each system respectively.
(a) (b)
Fig. 4. Density variation with time during MD simulation of (a) the second step (b) the third step.
Once the density of the system becomes stable as above, the next step is meaningful. The fourth step of MD is a production process and the fourth step of the trajectory has been written to the hard disk drive for later analysis. The size of a trajectory file of the fourth step of MD was about 250 GB. Due to the huge size of the trajectory file, we have installed additional 1 TB hard disk drive to the computer respectively. Moreover, swap memory of about 250 GB must be assigned to process a vector calculation successfully with ptraj, AMBER analysis program. The fourth step of MD Brownian dynamics, a Langevin dynamics without inertia has been performed. Generally, Brownian dynamics means without any outer thermal control, but we have used a very weak thermal control
P1-26 / J.-S. Kim
IMID 2009 DIGEST • in the production MD process to avoid an energy
leakage, affected by a numerical round-off error. Lager value of the temperature relaxation time of 5 ps has been applied for the weak coupling. Accordingly, we have performed statistical analysis to lower the numerical error.
By using mean director method, the rotational viscosity of two different kinds of the LC mixture has been obtained with the investigation of the sampling-time-width of the squared displacement of the mean director.
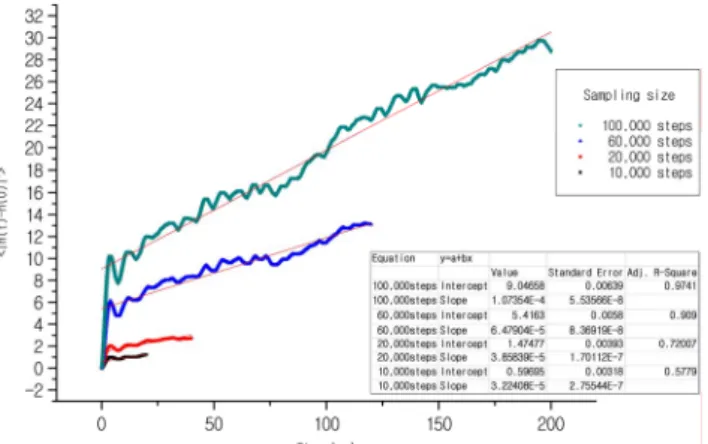
Figure 5 shows that as the length of the sampling-time becomes longer the slope of the line increase gradually and it has a tendency of the convergence of the slope. The slope of the squared displacements of the mean director with time has been calculated for each graph respectively using our own programming code.
Fig. 5. Slope of the DMSD as a function of time. The straight line shows the least squares fit.
At least 50 ps of time is needed to read the rotational viscosity, where 1 step of MD means 1 fs. Each point has been calculated from the slope of the line in the Figure 5.
Figure 6 shows comparison between the calculated value of rotational viscosity and the experimental findings previously obtained by Sudeshna DasGupta and Soumen Kumar Roy at various temperature and concentration of 5CB in the mixture.
Fig. 6. Variation of rotational viscosity with temperature for different concentrations of 5CB in the 5CB + 10CB system.
4. Summary
In summary, we have exhibited the mean director model by using molecular dynamics computer simulation. Rotational viscosity values of the 5CB + 10CB mixture in which mole fraction of 5CB is 0.3, 0.35 and 0.4 in respective nematic phases have been obtained from the Brownian fluctuation of the mean director of every nematic director with friction within the system by using Langevin dynamics technique.
5. References
1. Pierre Gilles de Gennes, J. Porst, The physics of Liquid Crystals, Clarendon Press, Oxford, U.K. (1994)
2. M. Smondyrev, George B. Loriot, Robert A. Pelcovits, Phys. Rev. Lett., 75, 2340 (1995)
3. Sten and Sarman, Physica A, 240, 160 (1997) 4. D. Demus, J. Goodby, G. W. Gray, H. W. Spiess, V.
Vill, Handbook of Liquid crystals, Vol. 2A, Wiley-VCH Press, Weinheim, 1998.
5. V. Zakharov, A. V. Komolkin, A. Maliniak, Phys. Rev. E, 59, 6802 (1999).
6. Satoru Kuwajima, Atsutaka Manabe, Chem. Phys. Lett., 332, 105 (2000).
7. D.L.Cheung, S.J. Clark, M.R.Wilson, Chem. Phys. Lett., 356, 140 (2002).
8. Sudeshna DasGupta, Soumen Kumar Roy, Phys. Lett. A, 306, 235 (2003).
9. M. Ilk Capar, E. Cebe, Chem. Phys. Lett., 407, 454 (2005).
10. J. Wang, R. M. Wolf, J.W. Caldwell, P. A. Kollamn & D. A. Case., J. Comput. Chem., 26, 114 (2005).