저작자표시-비영리-변경금지 2.0 대한민국 이용자는 아래의 조건을 따르는 경우에 한하여 자유롭게 l 이 저작물을 복제, 배포, 전송, 전시, 공연 및 방송할 수 있습니다. 다음과 같은 조건을 따라야 합니다: l 귀하는, 이 저작물의 재이용이나 배포의 경우, 이 저작물에 적용된 이용허락조건 을 명확하게 나타내어야 합니다. l 저작권자로부터 별도의 허가를 받으면 이러한 조건들은 적용되지 않습니다. 저작권법에 따른 이용자의 권리는 위의 내용에 의하여 영향을 받지 않습니다. 이것은 이용허락규약(Legal Code)을 이해하기 쉽게 요약한 것입니다. Disclaimer 저작자표시. 귀하는 원저작자를 표시하여야 합니다. 비영리. 귀하는 이 저작물을 영리 목적으로 이용할 수 없습니다. 변경금지. 귀하는 이 저작물을 개작, 변형 또는 가공할 수 없습니다.
Mast
er
'
sThesi
si
n
medi
ci
ne
Cl
i
ni
calappl
i
cat
i
on ofchr
omosomal
mi
cr
oar
r
ay as
af
i
r
st
-t
i
erchr
omosomeaber
r
at
i
on
t
estf
orpat
i
ent
swi
t
h devel
opment
al
del
ay andi
nt
el
l
ect
ualdi
sabi
l
i
t
y i
n
Kor
ea
Aj
ou Uni
ver
si
t
y Gr
aduat
eSchool
Maj
ori
n Medi
ci
ne
Depar
t
mentofMedi
calSci
ences
Cl
i
ni
calappl
i
cat
i
on ofchr
omosomal
mi
cr
oar
r
ay asaf
i
r
st
-t
i
er
chr
omosomeaber
r
at
i
on t
estf
or
pat
i
ent
swi
t
h devel
opment
aldel
ay
andi
nt
el
l
ect
ualdi
sabi
l
i
t
y i
n Kor
ea
Hyon J.Ki
m,Advi
sor
Seon-Yong J
eong,Advi
sor
Isubmi
tt
hi
st
hesi
sast
he
Mast
er
'
st
hesi
si
n medi
ci
ne.
Febr
uar
y 2017
Aj
ou Uni
ver
si
t
y Gr
aduat
eSchool
Maj
ori
n medi
ci
neDepar
t
mentofMedi
calSci
ences
Hyoungnam Lee
TheMast
er
'
st
hesi
sofHyoungnam Leei
n medi
ci
nei
s
her
eby appr
oved.
Thesi
sDef
enseCommi
t
t
eePr
esi
dent
Hyon J
.Ki
m
Thesi
sDef
enseCommi
t
t
eePr
esi
dent
Seon-Yong J
eong
Member Yoon-SokChung
Member Young BaeShon
Aj
ou Uni
ver
si
t
y Gr
aduat
eSchool
i
- ABSTRACT-
Clinical application of chromosomal microarray as a
first-tier chromosome aberration test for patients with
developmental delay and intellectual disability in Korea
Background: Chromosomal microarray (CMA) is a cytogenetic diagnostic test for chromosomal abnormalities in many diseases, which can provide the fast and accurate detection of copy-number variations (CNVs). International guidelines recommend specific approaches for the use of CMA as a first-tier clinical diagnostic test for patients with developmental disabilities or congenital anomalies. However, CMA is not used as a general diagnostic test in Korea because Korea’s insurance laws do not allow the use of CMA as a clinical diagnostic method. In this study, I aimed to investigate the clinical utility and limitations of CMA as a first-tier chromosome aberration test for patients with developmental delay (DD), intellectual disability (ID), and autism spectrum disorders (ASD) in Korea.Methods: Eighty-seven patients with neurodevelopmental disorders (16 ID, 70 DD and one ASD) and 32 patients with both congenital anomalies and symptoms of ID or DD revealed normal conventional G-banded karyotypes and were therefore referred by a clinical geneticist for further chromosomal analysis from December 2012 to July 2016. CMA was performed in a single clinical laboratory on a total of 182 samples from 119 patients and 63 family members of 37 patients. The clinical significance of CNVs and clinical features of patients and family members were studied.
ii
Results: The CMA results of 119 patients revealed 36 pathogenic CNVs (30.3%), 11 CNVs of variants of uncertain clinical significance (VOUS) (7.6%), 24 benign CNVs (17.6%), and no abnormalities in 53 patients
(44.5%). Among the 63 family members of 37 patients, the CMA results
revealed five pathogenic CNVs (8%), five VOUS (8%), 12 benign CNVs (19%), and no abnormalities in 41 cases (65%). In the 11 family members with abnormal phenotypes, the CMA results revealed five pathogenic CNVs (45.5%), two VOUS (18.2%), one benign CNV (9.1%), and three normal results (27.3%). The detection rate for pathogenic CNVs was 30.3% (36/119), which is higher than the average detection rate of previous reports.
Conclusions: In this study, pathogenic CNVs were detected in 36 patients (30.3%) and it was demonstrated that CMA is a very powerful tool with clinical diagnostic utility for patients with DD, ID, ASD, and congenital anomalies. However, there was a limitation of CMA results with VOUS for
interpretation for nine patients (7.6%). The significance of VOUS can be
improved by further testing through a familial studies and exome sequencing. Key Words: chromosomal microarray (CMA), copy-number variations (CNVs), congenital anomaly, developmental delay (DD), intellectual disability (ID)
iii
Table of Contents
Abstract ... i
Table of Contents ... iii
List of Figures ... iv
List of Tables ... iv
I. Introduction ... 1
II. Method A. Patients ... 3
B. CMA and analysis of chromosomal aberrations ... 4
III. Result A. CMA of patients ... 6
1. Pathogenic CNVs in neurodevelopmetal disorder patients 7 2. Pathogenic CNVs in congenital anomaly patients ... 11
B. CMA of patients’ family members ... 12
C. Chromosomal distribution of pathogenic CNVs ... 15
IV. Discussion ... 16
References ... 22
iv
List of Figures
Figure 1. Flow diagram of CMA test with patients and patients’ family
members ... 3
Figure 2. Distribution of pathogenic CNVs on chromosomes ... 15
List of Tables Table 1. The results of CMA patients ... 6
Table 2. Summary of pathogenic CNVs data for patients ... 8
Table 3. The results of CMA in patients’ family members ... 13
Table 4. Comparison of CMA and chromosomal genetic tests ... 14
Table 5. Comparison of CMA within family group and chromosomal genetic tests ... 17
Table 6. Comparison of recent chromosomal microarray detection rate in korea ... 18
1
I.
INTRODUCTION
DNA copy-number variations (CNVs) contribute significantly to the etiology of developmental delay (DD), intellectual disability (ID), autism spectrum disorders (ASD) and multiple congenital anomalies (MCA) [1-4].
Chromosomal microarray (CMA) is a powerful genetic test that provides a high-resolution, whole-genome examination of the chromosomes at once, allowing efficient and early diagnosis for the patients under suspicion of genetic diseases [5-6]. This technique overcomes the low-resolution (hard to evaluate under 5 Mb) problem of conventional G-banded karyotype analysis, and replaces the fluorescence in situ hybridization (FISH) test, which can analyze only targeted genes [7-11].
CMA has a high resolution [12]. According to the International Standard Cytogenomic Array (ISCA) Consortium and American College of Medical Genetics (ACMG) practice guidelines, CMA is widely used in the US and Europe as a first-tier clinical diagnostic test for patients with developmental disabilities or congenital anomalies [5,6]. Conventional G-banded karyotype analysis detects genomic imbalances only in only ~3% of patients with unexplained DD, ID, ASD, or MCA, whereas CMA detects genomic imbalances in 15–20% of these patients [3,5,13,14].
These international guidelines based in studies with cohorts in which CMA detects pathogenic CNVs, with an average diagnostic yield of 12.2% in 33 studies, including 21,698 patients with neurodevelopmental disorders or MCAs [5]. Though CMA has been introduced into clinical practices in Korea, it is not being used as a general diagnostic test because it is considerably more expensive than conventional G-banded karyotype testing and furthermore, it is not covered under the Korean National Health Insurance
2
Service. However, the availability of this technique has been applied in various diagnosis purpose of prenatal and postnatal, cancer study, DD/ID, ASD for suspected chromosomal abnormalities. Park et al. reported the clinical implementation of array CGH in total 4073 cases which include the
407 postnatal cases withDD, MCA and mental retardation and identified 34
deletion/duplication cases (8.3%, 34/407) [15]. Lee et al. showed the availability of array CGH that confirmed abnormal results in 26 patients out of 190 (13.7%) Korean patients with DD or ID combined with facial abnormalities of failure to thrive and Shin et al. suggested that CMA should be routinely performed in patients with unexplained DD, MR and ASD [4,14].
In this study, we evaluated the clinical availability and limitation of CMA as a first-tier chromosome aberration test for patients with DD, ID and congenital anomaly in Korea.
3
II.
METHODS
A. Patients
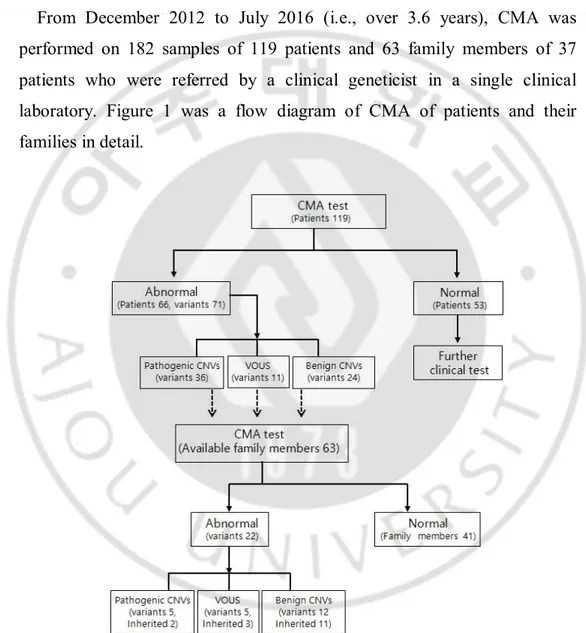
From December 2012 to July 2016 (i.e., over 3.6 years), CMA was performed on 182 samples of 119 patients and 63 family members of 37 patients who were referred by a clinical geneticist in a single clinical laboratory. Figure 1 was a flow diagram of CMA of patients and their families in detail.
Fig. 1. Flow diagram of CMA test with patients and patient’s family members
4
The patients were previously tested by conventional G-banded karyotype analysis, but no chromosomal abnormalities were detected in two university-affiliated hospitals and one local hospital: Konyang University Hospital, Gachon University Gil Medicine Center and Seoul Metropolitan Dongbu Hospital, respectively. A clinical geneticist evaluated the patients in depth, which included family members and pedigree.
Eighty-seven patients had neurodevelopmental disorders, with 16 ID, 70 DD and one ASD. Thirty-two patients had with both congenital anomalies and symptoms of ID or DD. Eleven family members were suspected of having abnormal features.
B. CMA and analysis of chromosomal aberrations
For each patient, peripheral blood in an EDTA bottle was sent to the clinical laboratory, Green Cross Genome Lab (Yongin, Gyeonggi, Korea). All samples were performed using a CytoScan 750K Array (Affimetrix, Santa Clara, CA, USA) and genome build Hg19 according to the manufacturer’s recommendations. The University of California at Santa Cruz Genome Browser (http://genome.ucsc.edu/) was used to map the locations of genomic alterations. All detected genomic changes were compared with the Database of Genomic Variants (DGV, http://dgv.tcag.ca/dgv/app/home), the Database of Chromosomal Imbalance and Phenotype in Humans using Ensembl Resources (DECIPHER; https://decipher.sanger.au.kr/), and the disease-causing genes referenced by Online Mendelian Inheritance in Man (OMIM, http://www.ncbi.nlm.nih.gov/omim).
According to the ISCA Consortium and ACMG practice guidelines, chromosomal aberrations were categorized as pathogenic CNVs, benign
5
CNVs, and variants of uncertain clinical significance (VOUS) [5] [16]. Pathogenic CNVs were defined as those that included a gene or part of a gene implicated in a known genetic disorder, overlapped with a region associated with a well-established syndrome, or represented deletions of large (>5 Mb) genomic regions. Parental and sibling testing was offered to aid further interpretation and classification.
6
III. RESULTS
A. The result of CMA in patients
One hundred and nineteen patients were tested in the 3.6 year period. We identified pathogenic CNVs in 36 patients (30.3%, 36/119); 22 (25.3%, 22/87) neurodevelopmental disorder patients and 14 (43.8%, 14/32) congenital anomaly patients, VOUS in nine patients (7.6%, 9/119), benign CNVs in 21 patients (17.6%, 21/119), and no abnormalities in the results of the rest (44.5%, 53/119) (Table 1).
Table 1. The result of CMA in patients
Abbreviation: ID, intellectual disability; DD, developmental display; ASD, autism spectrum disorder; CNVs, copy number variations; VOUS, variant of uncertain clinical significance.
7
1. Pathogenic CNVs in neurodevelopmental disorders’ patients A total of 87 neurodevelopmental disorder patients with 16 ID, 70 DD and one ASD were enrolled (Table 1).
Pathogenic CNVs were found in three patients with ID and 19 patients with DD. Eight patients had copy-number duplication, and 11 patients had copy-number deletion (Table 2).
Two patients had multiple pathogenic CNVs of two chromosomes; case 5 (2p36.3 to q37.3 duplication and 10p15.3 deletion) and case 11 (6q25.3 to q27 and 13q33.1 to p12 duplications).
The pathogenic CNVs were associated with known deletions and duplications in related human diseases in; case 14 (7q11.23, 1.5Mb deletion, Williams syndrome), case 15 (Xp21.2, 295 kb duplication, Duchenne muscular dystrophy) and case 20 (Xq22.2, 548 kb duplication, Pelizaeus– Merzbacher disease). Xq28 duplication (case 1) indicated that gain of MECP2 (methyl-CpG-binding protein 2, OMIM300005) gene was associated with infantile hypotonia, severe developmental delay, progressive neurological impairment, absence of speech, and proneness to infections. The deletion of 690 kb in 5q31.2q31.3 was detected in case 8. The NRG2 (neugegulin 2, OMIM603818) gene, is associated with neurodevelopment and developmental delay, is located in this region. Table 2 summarized the detailed descriptions of the results (Table 2).
8
Table 2. Summary of pathogenic CNVs data for patients (N =36 of 119 patients)
9
10
Table 2. Continued
Abbreviations: ID, intellectual disability; DD, developmental disability; S., syndrome; FTT, failure to thrive; BRRS, Bannayan Riley Ruvalcaba Syndrome.
11
2. Pathogenic CNVs in congenital anomaly patients
Pathogenic CNVs were found in 14 (43.8%, 14/32) congenital anomaly with symptom of ID or DD patients (Table 1).
Case 23 had a 55 Mb deletion at Xp22.33 to p11.21 and a 99 Mb gain at Xp11.21 to q28. These results suggest that the Xp11.21 region combined with a dicentric X chromosome may lead to a phenotype similar to Turner syndrome. Case 25 had a 2Mb deletion in 5q35.3q35.3, a region that includes NSD 1 (nuclear receptor-binding suvar, enhancer of zeste, and tirthorax domain protein 1, OMIM606681) gene, which is associated with Sotos syndrome. Case 26 had an 11 Mb deletion at Xp22.33 to p22.2 and a 12 Mb gain at Xq27.3 to q28, a region that includes SHOX (short-stature homeobox, OMIM 312865) gene, which is a well-known causative gene foe Léri–Weill syndrome.
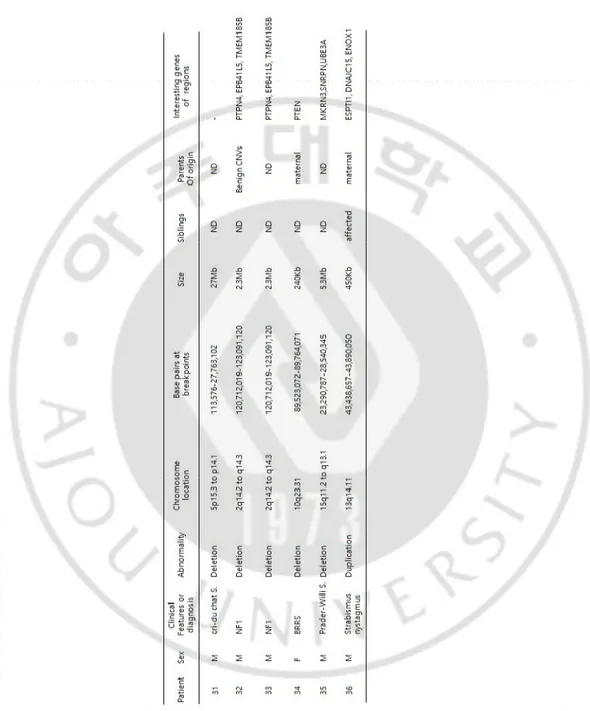
Case a 27 had a 1.7 Mb deletion at 7q11.23, a region that includes ELN (elastin, OMIM130160) LIMK1 (lim domain kinase 1, OMIM601329), EIF4H (eukaryotic translation initiation factor 4H, OMIM603431), LAT2 (linker for activation of T cells family member 2, OMIM605719), and CLIP2 (CAP-GLY domain-containing linker protein 2, OMIM60342) genes, which are associated with Williams syndrome. Case 28 had a 3 Mb deletion at 22q11.21, a deletion of this region that is a well-known cause of DiGeorge syndrome. Case 31 had a 27 Mb deletion at 5p15.3 to p14.1, a large deletion of this region which includes the critical region of Cri - du chat syndrome.
Two patients (cases 32 and 33) had the same clinical features and CMA results; a 2.3Mb deletion at 2q14.2 to q14.3 a region that includes PTPN4 (protein-tyrosine phosphatase nonreceptor type 4, OMOM176878) EPB41L5 (erythrocyte membrane protein band 4.1-like5, OMIM611730), GLI2 (GLI-kruppel family member 2, OMIM165230), TFCP2L1 (transcription factor
12
CP2-like 1, OMIM609785), CLASP1 (cytoplasmic linker-associated protein 1, OMIM605852), TSN (translin, OMIM600575), and INHBB (inhibin beat B, OMIM147390) genes, which it has been reported that deletion of CLI2 gene is related to a mild holoprosencephaly spectrum phenotype. Interestingly, Case 34 had a chromosomal abnormality of maternal origin, a 240 kb deletion at 10q23.31, a region that includes PTEN (phosphatase and tensin homolog, OMIM601728) and ATAD1 (atpase family AAA domain-containing member 1, OMIM614452) gene, which are associated with BRRS (Bannayan–Riley–Ruvalcaba syndrome). Case 35 had a 5.3Mb deletion at 15q11.2q13.1, a deletion of this region known to be a specific site of Prader– Willi and Angelman syndrome. Methylation PCR was performed to determine whether the patient had Prader–Willi or Angelman syndrome and was confirmed diagnosis of Prader–Willi syndrome.
CMA testing was offered to patients as their clinical features lacked sufficient information for diagnosis or they had a chromosomal variation was
suspected butnot be detected by conventional G-banded karyotype analysis.
B. CMA of patients’ family members
Patients were counseled with the significance of CMA results of pathogenic CNVs, VOUS and benign CNVs and then parental and sibling testing was offered to aid further interpretation and classification. In 63 family members of 37 patients, CMA showed pathogenic CNVs in five cases (8%, 5/63), VOUS in five cases (8%, 5/63), benign CNVs in 12 cases (19%, 12/63), and no abnormalities in 41 cases (65%, 41/63) (Table 3). Eleven family members showed abnormal phenotypes, which were associated with five pathogenic CNVs (45.5%, 5/11), two VOUS (18.2%, 2/11), and one benign CNVs (9.1%, 1/11), and no abnormalities were detected in three
13
cases (27.3%, 3/11) (Table 3).
Table 3. The result of CMA in patients’ family members
Abbreviation: CNVs, copy number variations; VOUS, variant of uncertain clinical significance.
All pathogenic CNVs were found in family members with abnormal phenotypes (Table 4). Three pathogenic CNVs were found to be the same or similar in patients’ CMA (Table 2, patient No. 24, 34, and 36). In family group 1 (Table 4), the patient (Table 2, patient No. 24) had a 140 kb deletion at 9p23 (9,387,329-9,528,560). Similarly, the patient’s sibling had a 140 kb deletion at 9p23 (9,387,329-9,525,866), a region that includes PTPRD (protein-tyrosine phosphatase receptor-type delta, OMIM601598) gene, the deletion of which has been reported to be associated with attention deficit hyperactivity disorder and restless leg syndrome.
In family group 2 (Table 4), the patient (Table 2, patient No.34) had a 240 kb deletion at 10q23.31 (89,523,072-89,764,071), and the patient’s mother had one at 10q23.31 (89,541,765-89,764,071), a region that includes PTEN gene, which is associated with BRRS. The patient and mother showed similar clinical phenotypes but the other family members had normal phenotypes and CMA results. In family group 3 (Table 4), not only the
14
patient (Table 2, patient No.36), but also the mother and sibling had the same clinical features of strabismus nystagmus, and the CMA showed similar results; the patient had a 450 kb duplication at 13q14.11 43,890,050), the sibling had a 450 kb duplication at 13q14.11 (43,438,657-43,890,050), and the mother had a 430 kb duplication at 13q14.11 (43,452,829-43,890,050). As a result of the CMA, the patient (Table 2, patient No. 26) in family group 4 (Table 4) was diagnosed with Leri-Weill syndrome and his younger brother was diagnosed with DiGeorge syndrome (Table 4).
Table 4. Comparison of CMA and chromosomal genetic tests
Abbreviations: CMA, chromosome microarray; FISH, fluorescence in situ hybridization.
15
C. Chromosome distribution of pathogenic CNVs
The pathogenic CNVs included 17 cases of long contiguous stretches of homozygosity of >5 Mb and one case (Table 2, patient No.22) of uniparental disomy at chromosomes 4 and 14 (Table 2). Genomic alterations occurred on all chromosomes except 19, 20, and Y. The changes were found most commonly (in order of frequency) on chromosomes 2, X, 3, 10, and 21 (Fig. 2).
Fig. 2. Distribution of pathogenic CNVs on chromosomes. Filled blue triangles, gains in patients; empty blue triangles, gains in family members with abnormal phenotypes; filled red triangles, losses in patients; empty red triangles, losses in family members with abnormal phenotypes. Stars show uniparental disomy.
16
IV. DISCUSSION
ID, DD and MCA are heterogeneous disorders, requiring proper genetic tests for accurate diagnosis, genetic counseling for patients and their families, and additional genetic tests for the family members [2,3,14]. There are many methods for detection of genomic changes [2,3,5]. Conventional G-banded karyotype analysis was the gold standard for detection of chromosomal abnormalities, but its resolution is limited (<5–6 Mb) [5]. Fluorescence in situ hybridization (FISH) was developed in the early 1980s. FISH uses probes labeled with a fluorescent dye to detect specific DNA sequences in chromosomes; FISH is used to detect structural chromosome abnormalities. Yet, uniparental disomy and germline mutations cannot be detected by FISH and it only detects abnormalities in the targeted regions corresponding to the probes used [5] (Table 5).
Conventional G-banded karyotype analysis only detects genomic imbalances in ~3% of patients with unexplained DD, ID, ASD, and cogeneital anomalies, whereas CMA detects genomic imbalances in 15–20% of these patients [3,5,17]. International guidelines (ISCA, ACMG) recommend specific approaches for the use of CMA as a first-tier genetic test in DD, ID, ASD, and cogeneital anomalies [6]. Despite the growing evidence for its diagnostic yield and cost effectiveness, CMA has not yet been implemented as a first-tier diagnostic test in Korea because it is not reimbursed by the Korean National Health Insurance Service.
Park et al. documented 4073 CMA tests with prenatal cases and 1007 postnatal cases, implemented array CGH in 407 patients with ID, MR, or MCA among postnatal cases, and confirmed chromosomal abnormalities of aneuploidy, deletion, and duplication in 34 patients (8.3%) in 2011 [15]. Lee
17
et al. performed array CGH in 190 Korean DD/ID patients, detected abnormal results in 26 out of 190 (13.7%), and reported that array CGH showed good clinical performance in 2013 [14]. Byeon et al. reported that the conventional G-banded karyotype method detected chromosomal abnormalities in only 9/87 patients, whereas array CGH revealed chromosome imbalances in 28/87 (32.2%) patients with DD, ID, dysmorphic face, or neurologic diseases, including epilepsy, in 2014 [13]. Shin et al. reported pathogenic CNVs in 15 patients (15.6%) among 96 patients with DD, MR or ASD, and insisted that CMA should be routinely performed for patients with DD, MR, or ASD [4] (Table 6).
Table 5. Comparison of CMA within family group and chromosomal genetic tests
18
Table 6. Comparison of recent chromosomal microarray detection rate in Korea
Abbreviations: ASD, autism spectrum disorder; DD, developmental display; MR, mental retardation; ID, intellectual disability; MCA, multiple congenital anomalies; CGH, comparative genomic hybridization; CNVs, copy number variations.
19
In this study, CMA analysis detected pathogenic CNVs in 43.8% (14/32) of cases of congenital anomaly and 25.3% (22/87) cases of neurodevelopmental disorder, which is higher than the average rate in previous reports [3,4,18,19]. The higher detection rate in this study than other reports could be explained by the high proportion of congenital anomaly among the participants. Focusing on neurodevelopmental disorders, the detection rate was 25.3%, which was similar to other reports [4,12,14,20]. Moreover, the type of array, genome coverage, and resolution could affect the detection rate. This study revealed 22 patients with a deletion, 9 patients with duplication, and 5 patients with duplication and deletion among pathogenic CNVs showing similarities with previous studies. Deletion occurs more often than random duplication in the genome and patients with duplications in their genome showed a milder phenotype than those with a deletion [3,18,21].
Pathogenic CNVs were randomly observed in all the chromosomes except 19, 20, and Y. However, duplication was observed mainly in the X chromosome (5/9, 55.6%) which differed from the study of Kaminsky et al. that reported duplication was common in chromosomes 3, 8, 16, and 17. The small number of participants could explain the difference [2].
This study included patients for whom the CMA results had been disclosed and recommendations had been suggested in genetic counseling. Then, CMA was performed on 63 family members of 37 patients and the results showed pathogenic CNVs in five members (8%). The comparison of CNVs found in patients and family members indicates the importance of family studies with phenotype examination and pedigree analysis for the accurate interpretation of CNVs found by the CMA test of the proband. The interpretation of VOUS can benefit from information on whether they are
20
inherited[5] [7]. The definition of a CNV is that at least 1 kb of DNA differs in copy number from a reference genome [6,21,22]. Not all CNVs have the same clinical significance; therefore, they are classified into pathogenic, benign, and VOUS [6,23]. However, with many genetic variants, there remains the possibility that a predicted pathogenic variant is not pathogenic and vice versa. In this study, the screening of one family (Table 4, family group 3) helped to interpret VOUS. The patient (Table 2, patient No. 36), sibling and mother had similar symptoms. CMA showed similar results of a ~450 kb duplication at 13q14.11, involving the genes ESPTI1, DNAJC15, and ENOX1; the patient’s father was normal. The clinical significance of this CNV has not yet been reported, but we interpreted it as pathogenic CNVs this family had due to an ophthalmopathy of abnormal phenotype.
In the US and Europe, CMA is a commonly used clinical diagnostic test with validated clinical utility, and is replacing conventional G-banded karyotype analysis and FISH [5,6]. Although CMA is a powerful diagnostic testing tool, screening using conventional G-banded karyotype analysis is more suitable for the detection of common aneuploidy [8,10]. FISH should be recommended for the diagnosis of well-described syndromes, such as Williams syndrome, and is a more cost-effective diagnostic test than CMA [24,25,26].
If a CMA result is normal, it does not mean that the genome is normal. Balanced translocation or inversions and low-level mosaic cases could be missed by CMA [27,28]. Therefore, a whole-chromosome analysis technique such as conventional G-banded karyotype analysis may be considered for patients with normal CMA results.
CMA has a limitation of interpretation with VOUS. The interpretation of VOUS can be helped by further testing of family study and exome
21
sequencing. Family study can inform whether it is inherited from a parent or it occurred de novo in the patient.
In summary, this experience demonstrates that CMA is a powerful cytogenetic test and chromosome aberration detection application for patients with congenital anomalies, DD, ID and ASD that enable the rapid diagnosis. I suggest that the CMA test should be covered by insurance and performed widely in clinical practices, which would help diagnose DD, ID, ASD and congenital anomaly patients accurately and provide the patients and their families with proper genetic counseling.
22
REFERENCES
1. Fan YS, Jayakar P, Zhu H, Barbouth D, Sacharow S, Morales A, et al. Detection of pathogenic gene copy number variations in patients with mental retardation by genomewide oligonucleotide array comparative genomic hybridization. Hum Mutat 2007;28:1124-32.
2. Kaminsky EB, Kaul V, Paschall J, Church DM, Bunke B, Kunig D, et al. An evidence-based approach to establish the functional and clinical significance of copy number variants in intellectual and developmental disabilities. Genet Med 2011;13:777-84.
3. Battaglia A, Doccini V, Bernardini L, Novelli A, Loddo S, Capalbo A, et al. Confirmation of chromosomal microarray as a first-tier clinical diagnostic test for individuals with developmental delay, intellectual disability, autism spectrum disorders and dysmorphic features. Eur J Paediatr Neurol 2013;17:589-99.
4. Shin S, Yu N, Choi JR, Jeong S, Lee KA. Routine chromosomal microarray analysis is necessary in Korean patients with unexplained developmental delay/mental retardation/autism spectrum disorder. Ann Lab Med 2015;35:510-8.
5. Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet 2010;86:749-64.
23
6. Kearney HM, Thorland EC, Brown KK, Quintero-Rivera F, South ST, Working Group of the American College of Medical Genetics Laboratory Quality Assurance C. American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet Med 2011;13:680-5.
7. Manning M, Hudgins L, Professional P, Guidelines C. Array-based technology and recommendations for utilization in medical genetics practice for detection of chromosomal abnormalities. Genet Med 2010;12:742-5.
8. de Ravel TJ, Devriendt K, Fryns JP, Vermeesch JR. What's new in karyotyping? The move towards array comparative genomic hybridisation (CGH). Eur J Pediatr 2007;166:637-43.
9. Armengol L, Nevado J, Serra-Juhe C, Plaja A, Mediano C, Garcia-Santiago FA, et al. Clinical utility of chromosomal microarray analysis in invasive prenatal diagnosis. Hum Genet 2012;131:513-23.
10. Coughlin CR, 2nd, Scharer GH, Shaikh TH. Clinical impact of copy number variation analysis using high-resolution microarray technologies: advantages, limitations and concerns. Genome Med 2012;4:80.
11. Ellison JW, Ravnan JB, Rosenfeld JA, Morton SA, Neill NJ, Williams MS, et al. Clinical utility of chromosomal microarray analysis. Pediatrics 2012;130:e1085-95.
24
12. Zilina O, Teek R, Tammur P, Kuuse K, Yakoreva M, Vaidla E, et al. Chromosomal microarray analysis as a first-tier clinical diagnostic test: Estonian experience. Mol Genet Genomic Med 2014;2:166-75.
13. Byeon JH, Shin E, Kim GH, Lee K, Hong YS, Lee JW, et al. Application of array-based comparative genomic hybridization to pediatric neurologic diseases. Yonsei Med J 2014;55:30-6.
14. Lee CG, Park SJ, Yun JN, Ko JM, Kim HJ, Yim SY, et al. Array-based comparative genomic hybridization in 190 Korean patients with developmental delay and/or intellectual disability: a single tertiary care university center study. Yonsei Med J 2013;54:1463-70.
15. Park SJ, Jung EH, Ryu RS, Kang HW, Ko JM, Kim HJ, et al. Clinical implementation of whole-genome array CGH as a first-tier test in 5080 pre and postnatal cases. Mol Cytogenet 2011;4:12.
16. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405-24.
17. Cho EH, Park BY, Cho JH, Kang YS. Comparing two diagnostic laboratory tests for several microdeletions causing mental retardation syndromes: multiplex ligation-dependent amplification vs fluorescent in situ hybridization. Korean J Lab Med 2009;29:71-6.
25
18. Shoukier M, Klein N, Auber B, Wickert J, Schroder J, Zoll B, et al. Array CGH in patients with developmental delay or intellectual disability: are there phenotypic clues to pathogenic copy number variants? Clin Genet 2013;83:53-65.
19. Moeschler JB, Shevell M, American Academy of Pediatrics Committee on G. Clinical genetic evaluation of the child with mental retardation or developmental delays. Pediatrics 2006;117:2304-16.
20. Vissers LE, de Ligt J, Gilissen C, Janssen I, Steehouwer M, de Vries P, et al. A de novo paradigm for mental retardation. Nat Genet 2010;42:1109-12.
21. Feuk L, Carson AR, Scherer SW. Structural variation in the human genome. Nat Rev Genet 2006;7:85-97.
22. Speicher MR and Carter NP. The new cytogenetics: blurring the boundaries with molecular biology. Nat Rev Genet 2005;6:782-92.
23. Kang JU and Koo SH. Clinical implementation of chromosomal microarray technology in prenatal diagnosis. (Review). Mol Med Rep 2012;6:1219-22.
24. Rauch A, Hoyer J, Guth S, Zweier C, Kraus C, Becker C, et al. Diagnostic yield of various genetic approaches in patients with unexplained developmental delay or mental retardation. Am J Med Genet A 2006;140:2063-74.
26
25. Coulter ME, Miller DT, Harris DJ, Hawley P, Picker J, Roberts AE, et al. Chromosomal microarray testing influences medical management. Genet Med 2011;13:770-6.
26. Trakadis Y and Shevell M. Microarray as a first genetic test in global developmental delay: a cost-effectiveness analysis. Dev Med Child Neurol 2011;53:994-9.
27. Bi W, Borgan C, Pursley AN, Hixson P, Shaw CA, Bacino CA, et al. Comparison of chromosome analysis and chromosomal microarray analysis: what is the value of chromosome analysis in today's genomic array era? Genet Med 2013;15:450-7.
28. Rehder CW, David KL, Hirsch B, Toriello HV, Wilson CM, Kearney HM. American College of Medical Genetics and Genomics: standards and guidelines for documenting suspected consanguinity as an incidental finding of genomic testing. Genet Med 2013;15:150-2.
27 -국문초록-
국내 발달지연과 정신지체 환자에 대한 우선적 검사로서
염색체 마이크로에레이의 임상적 적용에 대한 연구
이 형 남 아주대학교 대학원 의학과/의학전공 지도교수 김현주, 정선용 배경: 염색체 마이크로어레이 검사(CMA)는 염색체 전체를 고해상도로 빠르고 정확하게 복제수 변이(CNV)를 조사할 수 있는 세포유전학적 진단 검사법이다. 국제적 가이드라인은 발달 장애, 지적 장애, 자폐 범주성 장애, 선천성 기형 환자들에게 우선적 임상 진단 검사로서 적합한 검사법으로 추천하고 있다. 하지만, 국내에서는 건강 보험법상 임상진단 검사로 사용할 수 없어 CMA 검사가 일반적으로 사용되고 있지는 못한 실정이다. 본 연구에서는 CMA 검사가 발달 장애, 지적28 장애, 자폐 범주성 장애, 선천성 기형 등의 환자에게 우선적 진단 검사로서의 적 유용성과 한계점에 대해 연구하고자 하였다. 방법: 2012 년 12 월부터 2016 년 7 월 동안 염색체 이상이 의심되었으나 염색체 검사에서는 정상 핵형으로 관찰된 환자 119 명을 대상으로 하였다. 이중 87 명은 신경발달장애(발달지연 70 명, 지적장애 16 명, 자폐증 1 명)이고, 32 명은 발달지연이나 지적장애 증상을 가지고 있는 선천성 기형 환자였다. 환자 중 37 명은 가족 검사가 가능한 총 63 명의 환자가족들에 대해 CMA 검사를 실시하였으며, 검사결과를 바탕으로 유전상담을 시행하였다. 결과: 환자 119 명의 CMA 분석 결과 pathogenic CNV 36 명(30.3%), variants of uncertain clinical significance(VOUS) 11 명(7.6%), benign CNV 24 명(17.6%), 정상 53 명 (44.5%)으로 확인되었다. 환자 37 명의 가족들 63 명에 대한 CMA 결과는 pathogenic CNV 5 명 (8%), VOUS 5 명(8%), benign CNV 12 명(19%), 정상 41 명(65%)으로 분석되었다. 환자 가족 중 11 명은 비정상적 표현형을 보였으며 이들의 CMA 분석결과 pathogenic CNV 5 명(45.5%), VOUS 2 명(18.2%), benign CNV 1 명(9.1%), 정상 3 명(27%)로 pathogenic CNV 발견 빈도가 높았다. 환자 총 119 명중 pathogenic CNV 가 36 명으로
29 30.3%의 높은 빈도로 확인 되었다. 환자군에 따라 신경발달장애 환자 87 명중 22 명으로 25.3%로 확인되나 선천성 기형 환자 32 명중 14 명에서 확인되어 43.8%의 높은 비율을 확인할 수 있었다. 결론: 본 연구에서 신경발달장애 및 선천성 기형 환자에 대한 CMA 검사결과 30.3%에서 pathogenic CNVs 가 발견되었다. 비록 VOUS 결과의 해석적 한계점 등이 분명히 있지만 CMA 검사가 발달 장애, 지적 장애, 자폐 범주성 장애, 선천성 기형 등의 환자를 대상으로 한 우선적 염색체이상 검사법으로서 매우 유용하다는 것을 시사한다. 핵심어: 염색체 마이크로어레이, 복제수변이, 신경발달장애, 선천성 기형, 발달지연, 지적장애