저작자표시-비영리-변경금지 2.0 대한민국 이용자는 아래의 조건을 따르는 경우에 한하여 자유롭게 l 이 저작물을 복제, 배포, 전송, 전시, 공연 및 방송할 수 있습니다. 다음과 같은 조건을 따라야 합니다: l 귀하는, 이 저작물의 재이용이나 배포의 경우, 이 저작물에 적용된 이용허락조건 을 명확하게 나타내어야 합니다. l 저작권자로부터 별도의 허가를 받으면 이러한 조건들은 적용되지 않습니다. 저작권법에 따른 이용자의 권리는 위의 내용에 의하여 영향을 받지 않습니다. 이것은 이용허락규약(Legal Code)을 이해하기 쉽게 요약한 것입니다. Disclaimer 저작자표시. 귀하는 원저작자를 표시하여야 합니다. 비영리. 귀하는 이 저작물을 영리 목적으로 이용할 수 없습니다. 변경금지. 귀하는 이 저작물을 개작, 변형 또는 가공할 수 없습니다.
이학 박사학위 논문
고농도 포도당/
지방산 독성에 의한
I
NS-1베타세포사멸에서
Ni
cot
i
nami
de의 세포사멸 억제 기작
연구
아 주 대 학 교
대 학 원
의생명과학과
이 수 진
고농도 포도당/
지방산 독성에 의한
I
NS-1베타세포사멸에서
Ni
cot
i
nami
de의 세포사멸 억제 기작
연구
지도교수
강
엽
이 논문을 이학 박사학위 논문으로 제출함.
2013년
8월
아 주 대 학 교
대 학 원
의생명과학과
이 수 진
이수진의 이학 박사학위 논문을 인준함.
심사위원장
이
광
인
심 사 위 원
강
엽
인
심 사 위 원
이
수
환
인
심 사 위 원
최
성
이
인
심 사 위 원
김
진
규
인
아 주 대 학 교
대 학 원
2013년
6월 21일
-국문요약-고농도 포도당/지방산 독성에 의한 INS-1 베타세포사멸에서
Nicotinamide의 세포사멸 억제 기작 연구
높은 농도의 포도당과 지방산에 지속적으로 노출된 베타세포는 기능 이상에 이어 세포사멸이 유도되는데, 이는 제2형 당뇨병의 발병에 있어 중요한 병인이 다. Nicotinamide (NAM)은 nicotinamide adenine dinucleotide (NAD+) 대사에 관련된 중요한 전구물질로 TCA 회로를 통한 에너지 생산에 관여하며 NAD+를 기질로 사용하는 효소들의 활성을 억제한다. Streptozotocin (STZ)과 같은 베타 세포 독성물질에 노출된 상황에서 NAM의 처리는 베타세포의 양과 기능 감소를 예방한다고 알려져 있다. 또한, 고농도 포도당 (High glucose : HG)/지방산 (Plamitate : PA) 독성(Glucolipotoxicity)에 의한 베타세포사멸 (Apoptosis)에 서 NAM은 세포사멸을 억제하는 효과를 나타낸다. 본 연구에서 NAM을 통한 항 산화제로서 역할, NAD+의 전구체로서 역할 또는 NAD+ 소비 효소의 저해제로 서의 역할 중 어떤 역할에 의해 세포사멸이 억제되는지 알아보고 고농도 포도당/ 지방산 독성에 의한 세포사멸에서 NAD+ 대사 조절이 중요한 역할을 하는지 조 사하였다. 첫 번째, NAM이 항산화 작용으로 고농도 포도당/지방산 독성에 의한 세포사멸을 억제하는지 알기위해 여러 종류의 항산화제를 처리하여 세포사멸에 영향을 주는지 조사하였다. 항산화제인 N-acetylcystein (NAC), Reduced glutathione (GSH) 및 Mito-TEMPOL (Mito-T)를 처리했을 때, 고농도 포도 당/지방산 독성에 의한 세포사멸에 전혀 영향을 미치지 않았다. 두 번째, NAM을 통한 세포내 NAD+ 수준의 증가가 고농도 포도당/지방산 독성에 의한 베타세포 사멸에 영향을 주는지 그리고 NAD+ 합성 경로를 통한 NAD+ 공급이 고농도
포도당/지방산 독성에 의한 세포사멸에 연관되어 있는지 조사하였다. NAD+ de
novo 경로의 전구물질인 Kynurenin와 NAD+ salvage 경로의 전구물질인
nicotinamide mononucleotide (NMN) 또는 NAD+ 그 자체를 직접 세포외부에 서 처리했을 때 세포내 tNAD 양 (NAD+와 NADH)과 INS-1 세포의 viability 는 증가되지만 고농도 포도당/지방산 독성에 의한 세포사멸에는 영향을 미치지 않았다. Nicotinamide phospho ribosyl transferase (Nampt) 저해제로 알려진 FK866을 처리하거나 Nampt나 Nicotinamide mononucleotide adenylyl transferase (Nmnat)에 대한 siRNA로 각 유전자의 발현을 줄여 NAD+ salvage 경로를 저해했을 때, 세포내 tNAD+ 양은 감소하지만 고농도 포도당/지방산 독 성에 의한 베타세포사멸에서 NAM을 통한 세포사멸 억제 효과를 감소시키지 않 았다. 세 번째, NAM을 통한 NAD+ 소비 효소 활성의 저해가 고농도 포도당/지 방산 독성에 의한 세포사멸에 영향을 주는지 알기위해 NAD+ 소비 효소로 알려 진 poly(ADP-ribosyl) polymerase (PARP), cADP-ribose synthases (CD38), Sirtuin (SIRT)에 대한 저해제와 siRNA를 처리하였다. PARP 저해제인 3-AB 및 INH2BP를 처리하거나 PARP에 대한 siRNA를 처리하여 PARP 활성을 저해하 였을 때 고농도 포도당/지방산 독성에 의한 세포사멸에 영향을 미치지 않았다. CD38 발현이 저해되거나 과발현된 INS-1 세포 모두에서 고농도 포도당/지방산 독성에 의한 세포사멸에 영향을 미치지 않았다. 그러나 SIRT 활성화제인 resveratrol과 SIRT 저해제인 sirtinol을 처리했을 때 고농도 포도당/지방산 독성 에 의한 세포사멸이 resveratrol에 의해서는 오히려 증가하고 sirtinol에 의해서는 감소하였다. 마찬가지로 Sirt3, Sirt4에 대한 siRNA를 처리하여 Sirt3, Sitr4 발 현이 저해된 세포에서 고농도 포도당/지방산 독성에 의한 세포사멸이 억제되었 다. 반대로 Srit3, Sirt4가 과발현된 INS-1 세포에서는 고농도 포도당/지방산 독 성에 의한 세포사멸이 증대되었다. NAM은 고농도 포도당/지방산에 의해 유도되 는 소포체 스트레스 (ER stress) 지표분자인 P-JNK, CHOP, calnecxin을 감소 시키며, 반대로 고농도 포도당/지방산에 의해 감소된 생존신호로 알려진 P-Akt 신호는 회복시켰다. 위 결과들을 종합해 볼 때 NAM은 미토콘드리아 SIRT 활성
을 줄이는 저해제의 역할로 고농도 포도당/지방산 독성에 의한 세포사멸을 억제 한다고 제안할 수 있으며, 고농도 포도당/지방산을 처리하였을 때 미토콘드리아 내 대사 이상으로부터 유도되는 소포체 스트레스를 NAM의 처리를 통해 미토콘 드리아 대사 이상을 초기화함으로 세포사멸을 억제하였을 것으로 생각된다. 핵심어: Glucolipotoxicity, Nicotinamide, NAD+, Mitochondrial Sirtuins
차 례
국문 요약 ··· ⅰ 차 례 ··· ⅲ 그림 차례 ··· ⅴ 도표 차례 ··· ⅺ 약 어 ··· ⅻ Ⅰ. 서 론 ··· 1 A. 제2형 당뇨병 및 포도당/지방산 독성 ··· 1 B. 베타세포사멸 ··· 6 C. 미토콘드리아 기능 이상 - 베타세포 ··· 8 D. NAD 대사 ··· 91. NAD+ biology 와 NAD+ 합성 ··· 9
2. NAD+ 소비 효소 ··· 11 E. NAD+ 대사와 당뇨병 ··· 12 F. Niconinamide ··· 14 G. 미토콘드리아 sirtuin ··· 15 H. Glutamate dehydrogenase ··· 18 I. 연구목적 ··· 20 Ⅱ. 재료 및 방법 ··· 21 A. 재 료 ··· 21 B. 방 법 ··· 22 1. 세포주 및 세포 배양 ··· 22 2. MTT assay ··· 22 3. DNA fragmentation ··· 22 4. Western blotting ··· 22 5. 세포내 ATP량 측정 ··· 23
6. 세포내 tNAD량 측정 ··· 23 7. 미토콘드리아 분리 ··· 24 8. 형질주입 (Transfection) ··· 24 9. RT-PCR ··· 25 10. Plasmid vector 구축 ··· 25 가. pcCD38 ··· 25 나. pcNADK ··· 25
11. Glutamate dehydrogenase mutant plasmid vector 구축 ··· 26
가. ADP-ribosylation site mutant (C172A) ··· 26
나. GDH KR, KQ mutant ··· 26 12. 통계 ··· 27 Ⅲ. 결 과 ··· 32 A. 고농도 포도당/지방산 독성에 의한 INS-1 세포사멸에서 Nicotinamide의 세포사멸 억제 효과 ··· 32 B. STZ, 과산화수소, 사이토카인에 의한 세포사멸에서 Nicotinamide의 INS-1 베타세포사멸 억제 효과 ··· 35 C. Nicotinamide의 고농도 포도당/지방산 독성에 의한 베타세포사멸 억제 기작 ··· 37 1. 항산화제 : Nicotinamide를 통한 항산화제로서의 역할 ··· 37 2. NAD+ depletion ··· 41 3. NAD+ 보충 ··· 43 4. NAD+ salvage 경로 ··· 49 5. NAD+/NADH ··· 57 6. NADP+/NADPH ··· 59 7 . NAD+ 소비 효소 :Nicotinamide를 통한 NAD+ 소비 효소 저해제로서의 역할 ··· 61 가. PARP ··· 61
나. CD38 ··· 67 다. Situin ··· 69 D. Nicotinamide가 Mitochondiral Sirtuin의 저해제로서
고농도 포도당/지방산 독성에 의한 세포사멸에서 역할. ··· 77 1. 고농도 포도당/지방산에 의한 SIRT 발현 및 활성 ··· 77 2. 고농도 포도당/지방산에 의한 미토콘드리아 SIRT의 활성 ··· 80 E. ADP-리보실화, 아세틸화, 탈아세틸화에 의한 GDH의 활성 조절이 고농도 포도당/지방산에 의한 세포사멸에 미치는 영향 ··· 82 F. 고농도 포도당/지방산에 의한 신호전달에 Nicotinamide가 미치는 영향 ··· 87 IV. 고 찰 ··· 91 A. STZ, 과산화수소, 사이토카인에 의한 베타세포사멸에서 Nicotinamide의 세포사멸 억제 효과 ··· 92 B. 고농도 포도당/지방산 독성에 의한 베타세포사멸에서 항산화 작용이 미치는 영향 ··· 93 C. 고농도 포도당/지방산 독성에 의한 세포사멸에서 NAD+ 보충 및 NAD+ salvage 경로가 미치는 영향 ··· 94 D. 고농도 포도당/지방산 독성에 의한 세포사멸에서 NAD+ 소비에 관련된 효소들의 역할 ··· 95 E. 고농도 포도당/지방산 독성에 의한 세포사멸에서 미토콘드리아 SIRT의 역할 ··· 96 F. Nicotinamide를 통한 고농도 포도당/지방산 독성에 의한 세포사멸 억제 효과에 미토콘드리아 NAD+가 미치는 영향 ··· 98 G. 고농도 포도당/지방산 독성에 의한 베타세포사멸에서 ADP-리보실화, 아세틸화, 탈아세틸화를 통한 GDH 활성 조절이 미치는 영향 ··· 99 V. 결 론 ··· 102 참고 문헌 ··· 103
그림 차례
Fig.1. Islet β-cell failure and the natural history of T2D ··· 2 Fig. 2. Effect of glucose on lipid partitioning in the beta cell ··· 4 Fig. 3. Supplement of TCA cycle intermediates protects against high
glucose/palmitate-induced INS-1 beta cell death ··· 5 Fig. 4. Pathway to NAD+ synthesis ··· 10 Fig. 5. Network of mitochondrial sirtuins ··· 17 Fig.6. GDH activate stimulates the conversion of glutamate to
α-ketoglutarate ··· 18 Fig. 7. Construction of GDH mutant ··· 27 Fig. 8. NAM protects against HG/PA-induced INS-1 cell death ··· 33 Fig. 9. NAM protects against HG/PA-induced INS-1 cell death
- Western blotting ··· 34 Fig. 10. NAM protects against H2O2 or STZ-induced INS-1 cell death, but not against cytokine-induced cell death ··· 36 Fig. 11. Protective effect of NAM on HG/PA-induced INS-1 cell death is not attributable to its anti-oxidant activity ··· 39 Fig. 12. Protective effect of NAM on HG/PA-induced INS-1 cell death is not attributable to its anti-oxidant activity-ROS generation 40 Fig. 13. NAM prevents HG/PA-induced reduction of energy level ··· 42 Fig. 14. Supplementation of NAD+ does not protect HG/PA-induced
INS-1 cell death.-Exogenous NMN, NAD+ treatment ··· 45 Fig. 15. Supplementation of NAD+ does not protect HG/PA-induced
INS-1cell death.-Exogenous L-Kynurenine treatment ··· 46 Fig. 16. Supplementation of NAD+ increase intracellular tNAD level ··· 47 Fig. 17. Supplementation of NAD+ does not protect STZ-induced INS-1
cell death ··· 48 Fig. 18. Inhibition of NAD+ salvage pathway does not augment
HG/PA- induced INS-1 cell death ··· 51 Fig. 19. Inhibition of NAD+ salvage pathway decrease intracellular tNAD
level ··· 52 Fig. 20. Inhibotion of NAD+ salvage pathway does not augment
HG/PA-induced INS-1 cell death.-siRNA: siNampt, siNmnat ··· 54 Fig. 21. Agmentation of NAD+ salvage pathway does not protect
HG/PA-induced INS-1 cell death.-Visfatin overexpression ··· 55 Fig. 22. Inhibition of NAD+ salvage pathway protects STZ-induced
INS-1 cell ··· 56 Fig. 23. Decreased or increased of NAD+/NADH ratio does not protect
HG/PA -induced INS-1 cell death. - L-lactate, L-Asparagine treatment ··· 58 Fig. 24. Knockdown or overexpression of NADkinase (NADK) does not
protects HG/PA-induced INS-1 cell death ··· 60 Fig. 25. Inhibition of PARP does not protect HG/PA-induced INS-1 cell
death ··· 63 Fig. 26. PARP inhibitor 3-AB but not INH2BP increase intracellular tNAD
level ··· 64 Fig. 27. Inhibition of PARP does not protect HG/PA-induced INS-1 cell
death.-siRNA : siNampt, siNmnat ··· 65 Fig. 28. Pharmarcological inhibitor of PARP but not knock down of PARP
protects STZ-induced INS-1 cell death ··· 66 Fig. 29. Knockdown or overexpression of CD38 does not protect HG/PA -induced INS-1 cell death ··· 68 Fig. 30. Inhibition of SIRT protects against HG/PA-induced INS-1 cell
death ··· 71 Fig. 31. SIRT activator decrease intracellular tNAD level but SIRT inhibitor
does not increase intracellular tNAD level ··· 72 Fig. 32. Knockdown of Sirt3 or Sirt4 protects against HG/PA-induced
INS-1 cell death ··· 73 Fig. 33. Overexpression of Sirt3 or Sirt4 augment HG/PA-induced INS-1
cell death ··· 74 Fig. 34. Activation of SIRT protect STZ-induced INS-1 cell death ··· 76 Fig. 35. Expressioin of Sirtuins in the presence of HG/PA or in the presence of HG/PA and NAM ··· 78 Fig. 36. Pattern of Acetyl-lysine in HG/PA-treated INS-1 cell ··· 79 Fig. 37. Pattern of Acetyl-lysine in mitochondrial proetin or cytosolic
proteion of HG/PA-treated INS-1 cell ··· 81 Fig. 38. Construction of GDH mutant ··· 84 Fig. 39. Overexpression of GDH ADPr does not impact HG/PA-induced
INS-1 cell death ··· 85 Fig. 40. Overexpression of GDH KR form does not impact HG/PA-induced
INS-1 cell death. ··· 86 Fig. 41. Overexpression of GDH KQ form does not impact HG/PA-induced
INS-1 cell death ··· 87 Fig. 42. Inhibitory effect of nicotinamide on HG/PA-induced stress signal ··· 89 Fig. 43. Inhibitory effect of nicotinamide on HG/PA-induced stress signal (Graph) ··· 90
표 차례
Table. 1. Sirtuin localization and function ··· 15 Table. 2. Genbank number and nucleotide sequence of siRNA ··· 28 Table. 3. Nucleotide sequences of sets and reaction condition for PCR ···· 29 Table. 4. GDH mutant nucleotide sequences of sets ··· 30
STZ Streptozotocin
NAM Nicotinamide
NA Nicotinic acid
NR Nicotinamide ribose
NMN Nicotinamide mononucleotide
NAD+ Nicotinamide adenine dinucleotide
Nampt Nicotinamide phosphoryltransferase
Nmnat NMN adenylyltransferase
PARP Poly(ADP-ribose) polymerase
SIRT Sirtuin
GDH Glutamate dehydrogenase
Ⅰ. 서 론
A. 제2형 당뇨병 및 포도당/지방산 독성
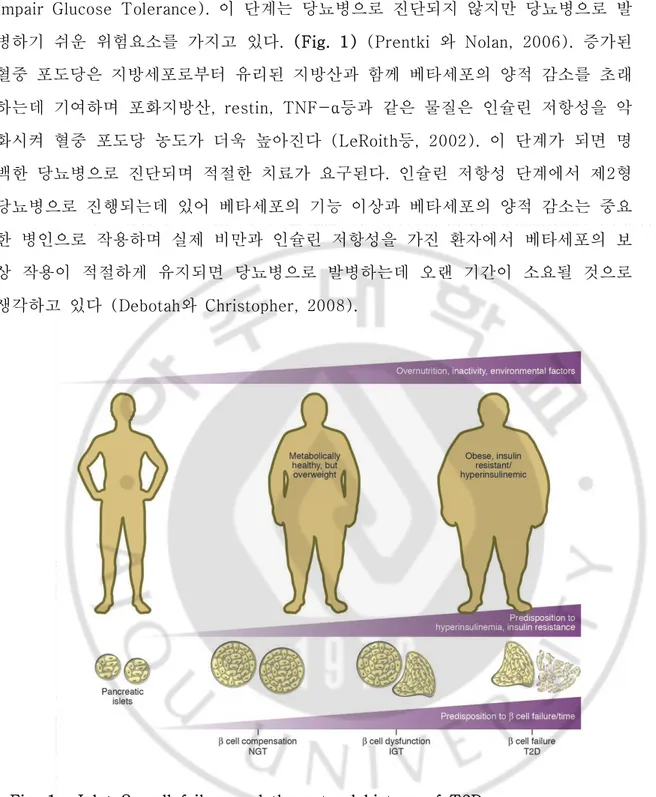
제2형 당뇨병은 간, 근육, 지방과 같은 인슐린 표적기관에서 인슐린의 작용 이 상 (인슐린 저항성 : Insulin Resistance)과 베타세포의 기능 이상에 따른 인슐린 분비 약화 (Beta-cell Dysfunction), 간에서 포도당 신 합성 증가, 인크레틴 호르몬 인 GLP-1의 조절과 분비 이상, 중추 신경계 균형의 변화와 같이 다양한 조직에서 비정상적인 조절에 의해 발병한다고 알려져 있으며, 위와 같은 비정상적인 조절에 의해 고혈당증 (Hyperglycemia), 고지혈증 (Hyperlipidemia)이 유지되는 질병이다 (Patti 등, 2010). 뿐만 아니라, 제2형 당뇨병의 발병과 진행에 있어 노화, 임신, 비 만과 같은 환경적 요인과 유전적 요인이 중요한 역할을 한다고 알려져 있다. 아직 까지 비정상적인 대사가 어떻게 유발되는지에 대한 기작은 정확히 알려져 있지 않 지만 인슐린이 제대로 분비되고 작용하지 않으면 고혈당증, 고지혈증과 같은 대사 조절에 이상이 유발된다. 고혈당, 고지혈 또는 두 가지 모두에 지속적으로 노출되면 베타세포에서 인슐린 분비가 약화되고 베타세포 손상이 일어나 혈중 인슐린이 감소 되며 혈중 포도당 및 지방산의 농도가 높아져 결국 다시 베타세포와 다양한 표적세 포에 영향을 주는 악순환을 통해 제2형 당뇨병이 더욱더 악화된다고 생각하고 있다 (Poitout 등, 2010). 비만으로 인해 유도된 인슐린 저항성은 간, 근육, 지방조직에서 인슐린 작용에 결함을 초래하고 포도당이 표적 기관으로 흡수되지 못해 혈중 포도당 농도가 높아 진다. 처음에는 이를 극복하고자 베타세포의 보상 작용을 통해 베타세포의 수가 증 가하며 인슐린 분비 또한 증가하여 정상적인 혈당을 유지할 수 있다 (NGT : Nomal Glucose Tolerance). 그러나 시간이 지날수록 인슐린 저항성에 대한 베타세 포의 보상 작용이 감소하게 되고, 베타세포의 기능 이상과 함께 인슐린 분비가 감 소하고 상대적 인슐린 농도가 부족하게 되어 혈중 포도당 농도는 증가된다 (IGT :Impair Glucose Tolerance). 이 단계는 당뇨병으로 진단되지 않지만 당뇨병으로 발 병하기 쉬운 위험요소를 가지고 있다. (Fig. 1) (Prentki 와 Nolan, 2006). 증가된 혈중 포도당은 지방세포로부터 유리된 지방산과 함께 베타세포의 양적 감소를 초래 하는데 기여하며 포화지방산, restin, TNF-α등과 같은 물질은 인슐린 저항성을 악 화시켜 혈중 포도당 농도가 더욱 높아진다 (LeRoith등, 2002). 이 단계가 되면 명 백한 당뇨병으로 진단되며 적절한 치료가 요구된다. 인슐린 저항성 단계에서 제2형 당뇨병으로 진행되는데 있어 베타세포의 기능 이상과 베타세포의 양적 감소는 중요 한 병인으로 작용하며 실제 비만과 인슐린 저항성을 가진 환자에서 베타세포의 보 상 작용이 적절하게 유지되면 당뇨병으로 발병하는데 오랜 기간이 소요될 것으로 생각하고 있다 (Debotah와 Christopher, 2008).
Fig. 1. Islet β-cell failure and the natural history of T2D. (Prentki 와 Nolan, 2006)
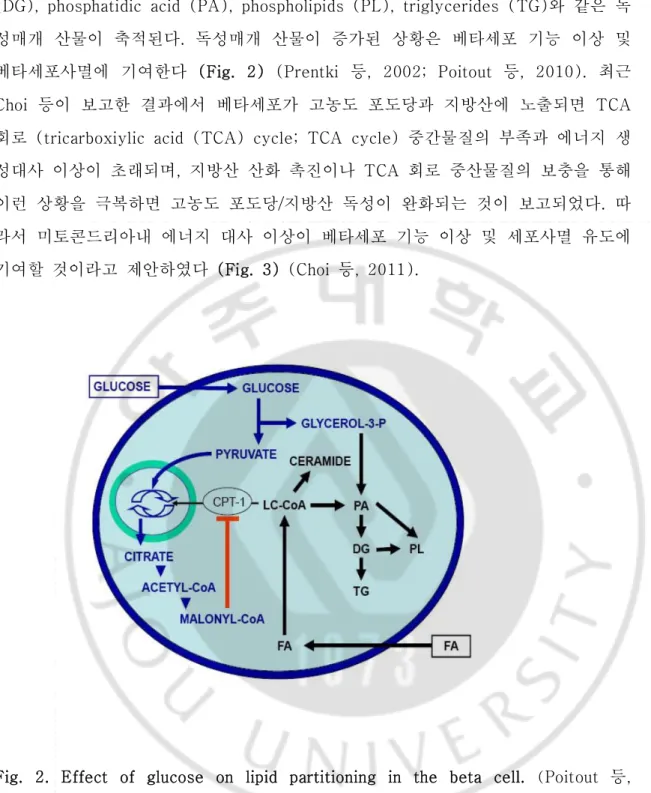
영양분이 과도한 상황 즉, 높은 농도의 포도당과 유리지방산의 증가는 활성산소 의 생성과, 대사과정의 이상, 세포내 칼슘의 증가, 소포체 스트레스 증가와 같은 다 양한 기작을 통해 인슐린의 분비가 감소하며 인슐린의 합성이 저해되어 결국 베타 세포사멸로 이어진다 (Chang-Chen등, 2008). 높은 농도의 포도당과 지방산을 이 용한 세포 수준의 실험을 통해 고농도의 포도당과 지방산 (Saturated fatty acid; Palmitate)은 베타세포의 기능 이상 및 세포사멸을 초래한다고 보고되었으며, 이는 각각 포도당 독성 (Glucotoxicity), 지방산 독성 (Lipotoxicity)이란 용어로 정의되 었다. 특히, 동시에 고농도 포도당과 지방산에 노출되면 베타세포의 기능 이상 및 세포사멸이 더욱 증가하며 이는 포도당/지방산 독성 (Glucolipotoxicity)이란 개념으 로 확립되었다 (Prentki 등, 1996). 마찬가지로 소도세포에 유리지방산을 장시간 노출하면 베타세포의 세포사멸이 일어나며, 이 조건에 포도당 농도를 높여주면 유 리지방산에 의한 세포사멸이 더욱 증가되는 것이 보고되었다 (El-Assaad 등, 2003). 그러나 베타세포에서 어떻게 포도당/지방산 독성이 일어나는지에 대한 기작 은 아직 정확하게 규명되어 있지 않다. Ceramide와 nitric oxide synthase의 활성에 의해 유도되는 일산화질소 (Nitric Oxide; NO)는 지방산에 의한 세포사멸에서 중요 한 매개인자로 알려져 있다. 또한 지방산에 의한 nPKC 활성, NF-κB 신호 활성이 지방산에 의한 세포사멸에 관여된다고 보고되었다 (Maedler 등, 2003; Okuyama 등, 2003; Eitel 등, 2003). 포도당/지방산 대사를 통한 과도한 활성산소 생산, 세포 질 내 칼슘 (Ca2+)의 증가, IRS/PI3K/Akt 신호 저해 또한 지방산에 의한 세포사멸 에 관여될 것이라 생각된다 (Piro 등, 2002; Choi 등, 2007; Lingohr등, 2003). 최 근 실험결과에 따르면 소포체 스트레스 (ER stress)의 활성이 포도당/지방산 독성 에 의한 베타세포사멸에 중요한 역할을 할 것이라 보고되었다 (Wang 등, 2005). 베타세포가 지속적으로 고농도 포도당과 지방산에 노출되면 포도당 대사로부터 만 들어진 malonyl-CoA양이 세포질 내에 증가하여 carnitin palmitoyltransferase (CPT)―1의 활성을 저해한다. 이로 인해 미토콘드리아 내로 long-chain acyl-CoA (LC-CoA) 유입이 감소하여 지방산의 베타산화 경로는 비활성화 되고, 상대적으로 지방합성 경로가 활성화 되는데 이 상황은 ceramide, diglycerides
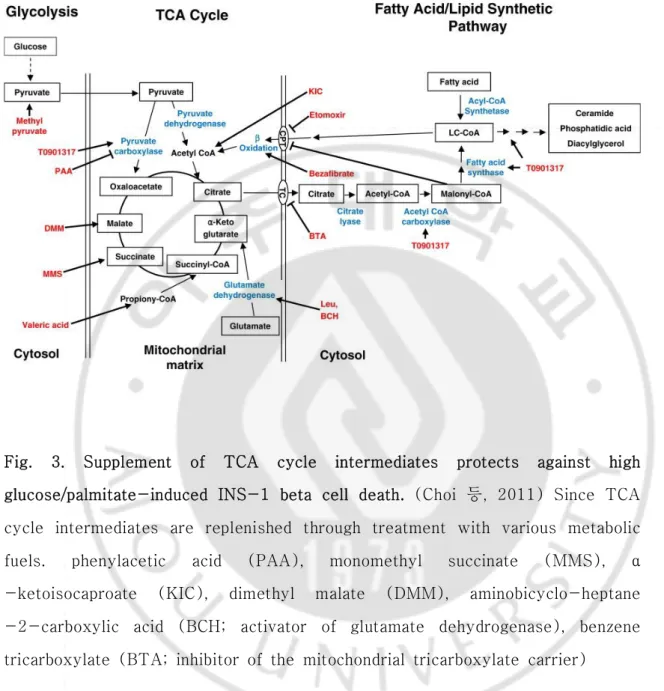
(DG), phosphatidic acid (PA), phospholipids (PL), triglycerides (TG)와 같은 독 성매개 산물이 축적된다. 독성매개 산물이 증가된 상황은 베타세포 기능 이상 및 베타세포사멸에 기여한다 (Fig. 2) (Prentki 등, 2002; Poitout 등, 2010). 최근 Choi 등이 보고한 결과에서 베타세포가 고농도 포도당과 지방산에 노출되면 TCA 회로 (tricarboxiylic acid (TCA) cycle; TCA cycle) 중간물질의 부족과 에너지 생 성대사 이상이 초래되며, 지방산 산화 촉진이나 TCA 회로 중산물질의 보충을 통해 이런 상황을 극복하면 고농도 포도당/지방산 독성이 완화되는 것이 보고되었다. 따 라서 미토콘드리아내 에너지 대사 이상이 베타세포 기능 이상 및 세포사멸 유도에 기여할 것이라고 제안하였다 (Fig. 3) (Choi 등, 2011).
Fig. 2. Effect of glucose on lipid partitioning in the beta cell. (Poitout 등, 2010) fatty acid (FA), carnitine-palmitoyl transferase-1 (CPT-1). long-chain acyl-CoA (LC-CoA), diglycerides (DG), phosphatidic acid (PA), phospholipids (PL), triglycerides (TG).
Fig. 3. Supplement of TCA cycle intermediates protects against high glucose/palmitate-induced INS-1 beta cell death. (Choi 등, 2011) Since TCA
cycle intermediates are replenished through treatment with various metabolic fuels. phenylacetic acid (PAA), monomethyl succinate (MMS), α -ketoisocaproate (KIC), dimethyl malate (DMM), aminobicyclo-heptane -2-carboxylic acid (BCH; activator of glutamate dehydrogenase), benzene tricarboxylate (BTA; inhibitor of the mitochondrial tricarboxylate carrier)
B. 베타세포사멸
당뇨병은 그 병인에 따라 제1형 당뇨병과 제2형 당뇨병으로 구분되는데, 제1형 당뇨병은 자가면역질환으로 인슐린을 분비하는 베타세포의 파괴로 인한 인슐린의 절대적인 부족으로 발병한다. 제2형 당뇨병은 제1형 당뇨병과 다르게 인슐린이 어 느 정도 분비됨에도 불구하고 인슐린 작용이 적절하게 이루어지지 못해 발병하는 질병으로서 발병 후에도 계속 당조절이 악화되어 혈당이 지속적으로 증가하는 진행 성 대사질환이다 (Hosker 등, 1989). 베타세포는 혈중 포도당 농도의 항상성을 유지하는데 중요한 기능을 담당하는 인슐린을 합성하고 분비하기 때문에, 베타세포의 양과 기능은 매우 엄격하게 조절 된다. 따라서 인슐린 분비에 중요한 역할을 하는 베타세포의 손실은 제1형 당뇨병 과 제2형 당뇨병에 있어 중요한 병인이라 생각할 수 있다. 베타세포의 운명은 다양 한 세포사멸 자극물질에 민감하다고 알려져 있으며, 베타세포의 사멸 (Apoptosis) 은 제1형 당뇨병이나 제2형 당뇨병에서 나타나는 공통적인 현상이다 (Mandrup-Poulsen, 2001). 제1형 당뇨병에서는 대식세포와 T-cell의 활성으로 인해 분비되는 염증성 사 이토카인 (Proinflammatory cytokine; TNF―α, INF―γ, IL―1β), 일산화질소, 활 성산소 (Reactive Oxygen Species; ROS) 또는 Fas 리간드에 의해 베타세포의 사 멸이 유도된다고 알려져 있다. 사이토카인은 세포내부의 신호전달, 세포내 ATP 양, 그리고 신호전달과정 중 단계별 인산화 (phosphorylation cascade)의 변화를 유도 하고 이로 인해 세포사멸에 관련된 유전자들의 발현이 증가하게 된다. NF-κB 신 호가 세포사멸을 유도하는 대표적인 신호전달로 생각되고 있다 (Kharroubi 등, 2004). 사이토카인에 의해 활성화된 NF-κB는 베타세포에 다양한 영향을 주는데, 첫 번째로 NF-κB의 활성은 inducible nitric oxide synthase (iNOS)의 발현을 증 가시켜 과도한 일산화질소의 생성을 촉진한다 (Darville, 1998). 두 번째로 베타세 포의 기능을 유지하는데 중요한 역할을 하는 것으로 알려진 duodenal homeobox-1 (PDX-1)의 발현을 감소시킨다 (Cardozo, 2001). 세 번째로 소포체 (EndopalsmicReticulum; ER)에 존재하는 칼슘 펌프로 알려진 Sarcoendoplasmic Reticulum Ca2+ ATPase type 2b (SERCA-2b)의 발현을 감소시킴으로 소포체내 칼슘 부족을 야기 하여 소포체 스트레스를 유도한다. 베타세포에 사이토카인을 처리하면 CHOP과 같 은 소포체 스트레스에 관련된 분자들이 유도된다는 결과가 보고되었으며 이들 분자 의 유도는 사이토카인에 의한 세포사멸에 중요한 역할을 한다고 생각하고 있다 (Oyadomari, 2002). 그러나 이와는 대조적인 결과로 소포체 스트레스를 줄여주는 약물인 PBA나 CHOP에 대한 siRNA를 베타세포에 처리했을 때 사이토카인에 의한 세포사멸에 영향을 미치지 않는 것에 대한 연구결과가 보고되었다 (Akerfeldt 등, 2008). 제2형 당뇨병에서는 유리지방산, 포도당, 설포닐요소제 등으로 인해 유도된 베 타세포의 세포사멸이 당뇨병 발병에 관여한다고 생각하고 있다. 고농도 포도당은 과도한 활성산소를 생성함으로써 산화적 스트레스 (Oxidative Stress)를 유발한다. 유리지방산에 의한 베타세포사멸 기작은 아직까지 정확히 밝혀져 있지 않지만, 세 포질 내에 fatty acyl-CoA의 축적, ceramide 신 합성의 증가, 미토콘드리아 (Mitochondria)에 존재하는 cytochrome C의 분비 그리고 protein kinase C (PKC) 의 활성은 protein kinase B (PKB)를 저해하여 베타세포사멸을 유도한다고 보고되 었다 (Kharroubi, 2004). 최근 연구결과에 의하면 유리지방산은 소포체 스트레스를 유도할 뿐 아니라 베타세포의 기능 이상과 세포사멸에 중요한 매개체로 생각되어지 고 있다 (Cnop 등, 2010). 제1형 당뇨병과 제2형 당뇨병에 의한 베타세포의 세포 사멸에서 세포사멸을 유도하는 자극은 다르지만 NF-κB 신호와 JNK 신호가 공통 적으로 활성화되는 특징을 가진다 (Cnop등, 2005). 당뇨병 연구에 사용되고 있는 Streptozotocin (STZ)은 베타세포에 특이적으로 반응하여 당뇨병을 유도하는 약제로써, 포도당과 유사한 구조를 가진 포도당 유도 체이다. STZ는 베타세포에 존재하는 포도당 수용체 (GLUT2)를 통해 베타세포내 로 들어가서 알킬화 작용으로 DNA나 단백질과 반응하여 각 물질의 변형을 유도하 여 독성을 나타내며 베타세포를 파괴하는 역할을 한다 (Lenzen, 2008). STZ에 의 해 유도되는 베타세포사멸 기작은 Okamoto 그룹에서 자세히 연구되었으며, STZ에
의해 손상된 DNA를 수리하기위해 poly(ADP-ribose) polymerase (PARP)라는 효 소가 활성화된다고 알려져 있다. 과도하게 활성화된 PARP에 의해 nicotinamide adenine dinucleotide (NAD+)가 소모되어 세포내 NAD+가 부족하게 되고 세포내 ATP 양 또한 감소하는데, 세포내 감소된 에너지에 의해 결국 베타세포가 사멸에 이르게 된다. 이를 Okamoto 모델이라고 제안되었다. (Okamoto와 Takasawa, 2002; Szkudelski, 2001). 이를 뒷받침하는 실험결과로 PARP 저해제를 처리하거나 PARP가 결손된 마우스에서는 STZ에 의한 세포사멸의 저해가 보고되었다 (Charron와 Bonner, 1999).
C. 베타세포에서의 미토콘드리아 기능 이상
베타세포에서 혈중 포도당 농도를 인지하고 대사시켜 인슐린 분비 및 합성을 하 는데 있어 미토콘드리아는 매우 중요한 역할을 한다. 포도당이 수용체를 통해 베타 세포 안으로 들어가면 해당 작용 (Glycolysis)을 통해 피루브산 (Pyruvate)이 만들 어 지고, 피루브산은 미토콘드리아로 들어가 acetyl-CoA로 바뀌어 TCA 회로를 거친다. TCA 회로를 통해 만들어진 NADH와 FADH2는 전자전달계로 전달되어 ATP
를 만든다. 전자전달계는 5개의 복합체로 이루어져 있으며 복합체 1은 NADH로부 터 하나의 전자를 전달받고 복합체 2는 FADH2로부터 전자를 조효소-Q로 옮겨준 다. 복합체 4에서는 산소를 소비하여 물을 생산하고 복합체 5에서는 복합체 4의 기 능과 연관하여 ADP로부터 ATP를 생산한다. 설명한 과정을 거쳐 포도당으로부터 생산된 ATP는 베타세포에서 인슐린 분비와 합성에 중요한 역할을 한다. 그러므로 베타세포에서 미토콘드리아의 기능 이상은 ATP 생산에 문제를 초래하고 베타세포 의 기능 이상을 유발하며 이는 결국 베타세포의 사멸까지 유도할 수 있다 (Supale 등, 2012). 활성산소는 정상적인 대사과정에서 전자전달계를 통한 부산물로 자연스럽게 생 성되며, 생성된 활성산소는 세포내 또는 미토콘드리아 항산화효소에 의해 제거된다. 그러나 에너지가 과도하게 요구되는 상황이나 스트레스가 유도된 상황에서는 전자
전달계의 이상으로 미토콘드리아 내에 자유라디칼 (free-radical)이 과도하게 증가 하고 ATP 생산이 감소되며 세포내에 활성산소가 축적된다. 산화물질에 직접적인 노출이나 포도당/지방산 독성에 의해 이차적인 반응으로 유도된 산화적 스트레스는 베타세포의 기능을 약화시킨다고 보고되었다 (Robertson 등, 2004).
미토콘드리아 DNA는 13개의 펩타이드, 22개의 tRNA, 2개의 rRNA의 유전정보 를 담고 있는 37개의 유전자로 이루어져 있다. 대부분의 미토콘드리아 단백질은 핵 으로부터 번역되고 만들어져 미토콘드리아로 이동하지만 전자전달에 관련된 몇몇 구성체는 미토콘드리아에서 번역되어 만들어 진다. 따라서 미토콘드리아 DNA의 변 이는 미토콘드리아의 기능에 문제를 야기할 수 있다. 뿐만 아니라, 미토콘드리아 DNA의 변이는 칼슘 항상성 조절에 문제를 야기하며 미토콘드리아의 대사 이상을 유발하여 과도한 활성산소를 생성한다고 보고되었다 (de Andrade 등, 2006). 미토 콘드리아에 존재하는 전사인자 A (Transcription factor A; Tfam)가 결손된 마우스 에서 산화적 인산화의 이상이 유발되며 포도당 자극 인슐린 분비가 감소됨이 보고 되었다 (Silva 등, 2000).
D. NAD 대사
1. NAD+ biology 와 NAD+ 합성
1930년대에 Otto Warburg에 의해 NAD+의 기능과 생화학적인 조절이 잘 규명 되었다 (Warburg와 Christian, 1936). NAD+는 NADP+, NADH, NADPH가 탈인 산화 되거나 환원된 형태로 대사의 산화환원 균형에서 중요한 역할을 하는 조효소 로 잘 알려져 있다. 좀 더 자세히 설명하면 포도당과 지방산은 산화되어 에너지원 으로 이용되는데 지방산 산화, 해당 작용, TCA 회로를 통해 포도당 및 지방산의 환원력은 NAD+에 전달되고 환원된 형태의 NADH를 만든다. NADH를 통해 운반 된 전자는 전자전달계로 전달되어 산화적 인산화 과정을 통해 ATP가 생성된다. 이 과정에서 산화형태인 NAD+, 환원형태인 NADH 형태를 제공함으로 탈수소효소 (Dehydrogenase) 반응을 촉매하며 세포의 에너지를 생산하는데 중요한 역할을 한
다 (Houtkooper 등, 2010).
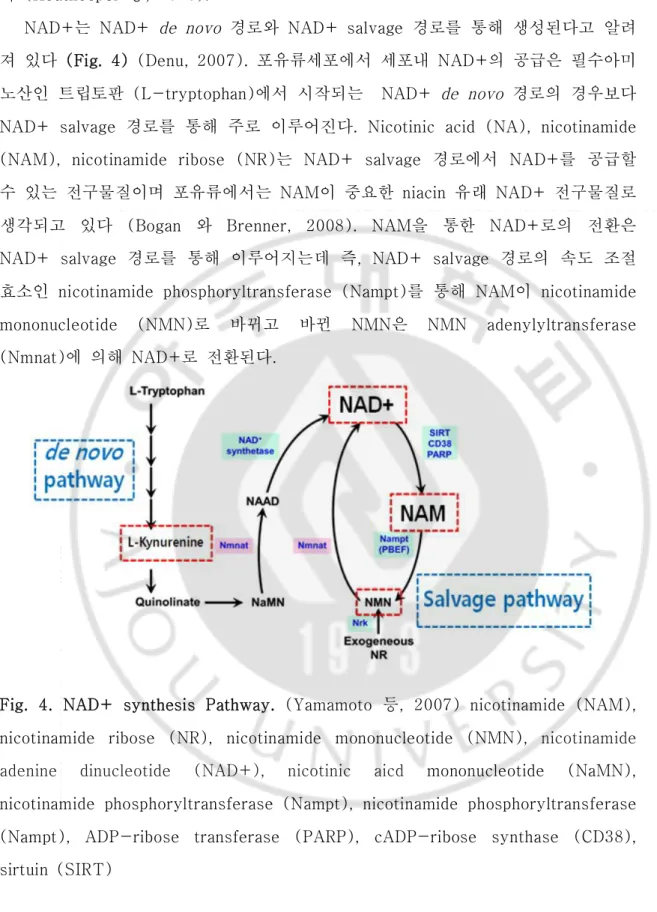
NAD+는 NAD+ de novo 경로와 NAD+ salvage 경로를 통해 생성된다고 알려 져 있다 (Fig. 4) (Denu, 2007). 포유류세포에서 세포내 NAD+의 공급은 필수아미 노산인 트립토판 (L-tryptophan)에서 시작되는 NAD+ de novo 경로의 경우보다 NAD+ salvage 경로를 통해 주로 이루어진다. Nicotinic acid (NA), nicotinamide (NAM), nicotinamide ribose (NR)는 NAD+ salvage 경로에서 NAD+를 공급할
수 있는 전구물질이며 포유류에서는 NAM이 중요한 niacin 유래 NAD+ 전구물질로
생각되고 있다 (Bogan 와 Brenner, 2008). NAM을 통한 NAD+로의 전환은 NAD+ salvage 경로를 통해 이루어지는데 즉, NAD+ salvage 경로의 속도 조절 효소인 nicotinamide phosphoryltransferase (Nampt)를 통해 NAM이 nicotinamide mononucleotide (NMN)로 바뀌고 바뀐 NMN은 NMN adenylyltransferase (Nmnat)에 의해 NAD+로 전환된다.
Fig. 4. NAD+ synthesis Pathway. (Yamamoto 등, 2007) nicotinamide (NAM), nicotinamide ribose (NR), nicotinamide mononucleotide (NMN), nicotinamide adenine dinucleotide (NAD+), nicotinic aicd mononucleotide (NaMN), nicotinamide phosphoryltransferase (Nampt), nicotinamide phosphoryltransferase (Nampt), ADP-ribose transferase (PARP), cADP-ribose synthase (CD38), sirtuin (SIRT)
2. NAD+ 소비 효소
NAD+는 앞에서 언급한 바와 같이 수소전달반응에서 조효소로의 역할 뿐만 아 니라 ADP-ribose의 공여체로 사용되어 NAD+가 소비되는 반응에도 중요한 역할 을 한다. NAD+를 기질로 사용하여 NAD+ 소비에 관여하는 효소로는 ADP-ribose transferase (PARP), cADP-ribose synthase (CD38, CD157), sirtuin (SIRT) 등 이 대표적으로 알려져 있다 (Fig. 4). 이 효소들의 경우 간접적으로 NAD+의 생리 학적 효과에도 영향을 줄 뿐만 아니라 에너지 대사, 세포의 생존, 노화에 중요한 영 향을 미친다. 또한 이 효소의 활성은 NAD+의 생리학적인 양의 변화에 따라 조절 되는 것으로 추정된다 (Houtkooper 등, 2010). 최근 10년간 연구를 통해 NAD+ 소비 효소이며 대사 조절인자로의 역할을 가지고 있는 sirtuin과 같은 단백질이 알 려짐으로써 NAD+에 대한 새로운 역할이나 기능에 대한 연구가 새로이 조명되었으 며 제2형 당뇨병이나 암과 질병에서 대사 조절인자로서 NAD+의 역할의 중요성이 대두되고 있다.
PARP는 핵에 존재하는 효소로 chromatin 구조나 DNA 대사에 관여하는 표적 단백질에 NAD+를 기질로 사용하여 ADP-ribosyl group을 붙여주는 역할을 한다.
PARP의 활성이 NAD+ 분해 활성에 중요한 역할을 한다는 것이 알려져 있으며
PARP의 활성 증가는 세포내 NAD+ 수준에 영향을 미친다 (Schreiber 등, 2006). CD38과 CD157은 NAD+를 기질로 사용하여 cADP-ribose와 같은 이차전령 (Second messenger)을 만들어내고 만들어진 cADP-ribose는 칼슘 이동에 중요한 역할을 한다고 알려졌다 (Lee, 2006). 하지만 cADP-ribose을 합성하는 기능보다 는 NAD+를 분해하는 기능이 이 효소의 주된 기능으로 생각되어지는데 그 이유는 CD38이 CADRP 한 분자를 만들어 내는데 100 분자 NAD+를 분해시켜 소모하기 때문이다. NAD+ 수준을 조절하는 분자로서 기능을 알기위해 CD38이 결손된 마우 스의 조직에서 NAD+ 양을 측정해 봤을 때 높게는 20배 이상 NAD+ 양이 증가하 였다. 따라서 CD38은 세포내 NAD+ 수준을 조절하는데 중요한 조절인자로 추정된 다 (Aksoy 등, 2006).
를 제거하는 활성을 가진다. 일반적으로 히스톤 (Histone) 단백질이나 다른 단백질 의 리신 (Lysine)기에 아세틸화 (acetylation) 변형을 유도한다고 알려졌으나 노화 와 관련된 유전자의 전사단계에서의 유전자 발현조절 (Transcriptional silencing)을 통해 수명 연장과 관련이 있다는 보고에 의해 각광받기 시작했다 (Kaeberlein 등, 1999). 뿐만 아니라, 물질대사를 인지하는 센서로서 대사적 상태 (Metabolic state)에 따라 많은 유전자 발현을 조절하는 매개인자로써 역할이 보고되었다 (Guarente, 2000). Sirt1은 굶었거나, 운동을 하거나, 포도당 이용이 부족한 상황과 같은 에너지 스트레스 상황 즉, 세포내 NAD+ 수준이 증가되는 상황에서 활성화되 며 활성화된 Sirt1은 peroxisome proliferator-activated receptor γ (PPARγ), PPAR γ coactivator-1α (PGC-1α)과 같은 대사 조절에 관련된 전사인자들의 유 전자 발현을 증가시킴으로 세포내 대사를 조절한다. 간에서 굶은 상태와 정상상태 를 비교했을 때, 굶은 상태에서 NAD+양이 50% 증가했으며 이와 함께 Sirt1 활성 을 통한 표적 단백질의 탈아세틸화와 PGC-1의 활성의 증가가 보고되었다 (Rodgers 등, 2005).
E. NAD+ 대사와 당뇨병
SIRT 활성화를 통한 에너지 대사활성의 증가가 당뇨병이나 대사증후군을 완화 와 같은 연구결과들이 최근 많이 보고되었다. 따라서 이들 질병의 예방이나 치료를 위해 SIRT의 활성을 증가시키기 위한 방법이 다양하게 연구되었으며, SIRT의 활성 에 중요한 역할을 하는 NAD+ 즉, 세포내 NAD+ 농도를 증가시키는 방법을 통해 SIRT 활성 조절이 제안되었다.첫 번째로 NR, NA, NAM와 같은 NAD+의 전구체 중 하나를 처리하여 세포내 NAD+ 수준을 증가시키는 것이다. 쥐 소도세포에 고농도 포도당 또는 유리지방산 을 처리하여 세포사멸을 유도한 조건에 5 mM NAM를 처리하면 세포사멸을 억제 하는 효과가 있다 (Piro 등, 2002). 근육세포 (Myotube)에서도 과산화수소 (H2O2) 를 처리해 DNA 손상을 야기하면 과도한 PARP의 활성으로 인해 세포내 NAD+ 양
의 감소와 함께 Sirt1의 활성이 감소하는데, 이 조건에 세포배양 배지에 직접적으로 NAD+를 첨가해 주거나 NAD+ 합성에 관련된 효소들을 과발현시키면 세포의 생존 과 SIRT의 활성이 회복되었다 (Pillai 등, 2005). 임상 실험 결과에서도 마찬가지로 제1형 당뇨병 환자에 NAM을 처리하였을 때 베타세포에 주는 손상을 저해함으로 당뇨병의 발병을 예방하거나 지연시키는 효과가 있음이 보고되었다 (Gale 등, 2004). 특히, 최근 연구결과에 의하면 식이 또는 노화에 의한 당뇨병 모델 쥐에 NAD+의 중요한 전구물질인 NMN을 처리하였을 때 식이 또는 노화에 의해 감소되 었던 NAD+ 양이 증가하였으며, 내당능장애가 개선되었다 (Yoshino 등, 2011). 두 번째로 SIRT를 제외한 PARP, CD38과 같이 NAD+를 소비하는 효소를 선택 적으로 저해함으로 세포내 NAD+의 양을 증가시켜 SIRT의 활성을 증가시키는 것 이다. CD38이 결손된 마우스에서 식이를 통한 비만이 억제되는 효과가 나타났는데 이는 일반적으로 잘 알려진 NAD+ 의존적인 Sirt1-PGC 1α 경로를 통해 에너지 소비가 증대됨으로 인해 비만이 억제된 것으로 추측된다 (Barbosa 등, 2007). 또한 CD38의 저해제를 처리하면 세포내 NAD+ 양과 SIRT의 활성 조절을 통해 전반적 인 단백질의 아세틸화를 조절하여 포도당, 지방 대사를 개선한다고 보고되었다 (Escande 등, 2013). PARP1이 결손된 마우스에서 NAD+양이 증가하며 Sirt1의 활성을 통한 대사 활성 증가가 보고되었다 (Bai 등, 2011). 따라서 CD38이나 PARP의 선택적인 억제제는 아마도 NAD+ 의존 효소인 Sirt1의 활성을 증가시킬 것으로 기대할 수 있다.
마지막으로 NAD+ salvage 경로에서 속도-조절 효소로 알려진 Nampt의 발현 조절을 통해 NAD+ 수준을 증가시키는 것이다. Nampt 양이 감소한 암컷 쥐에서 세포내 NMN의 양이 감소하고 포도당 자극 인슐린 분비가 약화되어 있음이 보고되 었다 (Revollo 등, 2007). 그러므로 Nampt의 발현 양을 증가시켜주거나 NMN 양 의 보충은 당뇨병 개선에 도움을 줄 수 있다. NAD+ 대사조절을 통한 SIRT의 활성 증가는 미토콘드리아 활성 증가로 이어진 다고 생각되며, 대사질환에서 유익한 효과를 나타냄으로 SIRT의 활성을 증가시킬 수 있는 약물의 개발이나 NAM/NMN/NAD+의 합성 경로를 조절하는 방법을 통해
대사질환의 치료와 예방을 위한 대안이 될 수 있다.
F. Nicotinamide
NAM는 vitamin B3의 amide 형태로 NAD+의 전구물질로 알려져 있다 (Li 등, 2004). 또한, NAM는 세포에서 NAD+로 전환되어 TCA 회로를 통한 에너지 대사 에 관여하며 미토콘드리아 전자전달계를 통한 ATP 생산에 관여하며 PARP 활성을 통해 DNA 합성과 수리에도 관여한다 (Lin과 Guarente, 2003; Hageman 과 Stierum, 2001). 또한 NAM은 세포사멸에도 관여하는데, NAM을 허혈성 조직에 처 리하면 손상 받은 조직에서 NAD+ 수준을 유지함으로써 세포사멸의 억제가 보고되 었다 (Sadanaga 등, 2003). NAM을 통한 세포내 에너지 공급과 세포 기능 유지는 미토콘드리아의 기능과 긴밀하게 연결되어 있다. 또한, NAM은 미토콘드리아의 막전위 (Membrane potential)를 유지함으로 세포의 대사 항상성을 유지하는데도 중요한 역할을 할 수 있다. 산화적 스트레스와 같은 상황에서는 미토콘드리아 permeability transition pore (PTP)가 열리고 NAD+ 부족이 야기되며 세포사멸이 일어나는데 이런 상황에 NAM을 처리하여 NAD+ 수준을 증가시키면 세포사멸을 억제할 수 있다고 보고되 었다 (Du 등, 2003). 또한 NAM은 Akt 신호전달의 활성을 통해 BAD를 인산화시 킴으로써 미토콘드리아 막의 탈분극화 (Membrane depolarization)를 예방하고 미 토콘드리아로부터 cytochrome C의 분비를 저해한다 (Chong 등, 2005).
NAM은 NAD+ 소비 효소로 알려진 PARP, CD38, SIRT의 저해제로서 이들 효 소의 효소활성을 저해할 수 있다 (Rankin 등, 1989; Sauve 등, 1998; Sauve 등, 2005). 자유라디칼에 의해 손상 받은 lymphocyte에서 NAM을 처리하면 과도한 PARP의 활성이 제한됨으로 인해 NAD+의 소비를 줄여 세포사멸이 억제된다 (Tronov 와 Konstantinov, 2000). NAM은 SIRT의 활성을 감소시키시는 억제제로 서의 역할도 보고되었는데, 식이를 제한한 효모에 NAM을 처리하면 Sir2와 pyrazimidase /nicotinamidase 1 (PNC1)의 발현 증가를 통한 수명 연장 효과가 사
Sirtuin Class Localization Activity Target
SIRT1 Ⅰ Nucleas, cytosol
deacetylation PGC-1α, FOXO1, FOXO3, p53, NF-κB, HIF1α, LXR, FXR and more
SIRT2 Ⅰ Cytosol deacetylation Tubulin, PEPCK, FOXO1, PAR3
SIRT3 Ⅰ Mitochondria deacetylation LCAD, HMGCS2, GDH, SOD2, IDH2, OXPHOS complex
SIRT4 Ⅱ Mitochondria ADP-ribosylation GDH SIRT5 Ⅲ Mitochondria deacetylation,
demalonylation desuccinylation
CPS1
SIRT6 Ⅳ Nucleus deacetylation ADP-ribosylation
H3K9, H3K56 SIRT7 Ⅳ Nucleolus Unknowm Unkonown
라지는 것이 보고되었다 (Anderson 등, 2003; Bitterman, 2002). 이는 NAM이 Sir2의 억제제로서의 역할을 통해 수명 연장 효과가 억제되는 것으로 추측할 수 있다.
G. 미토콘드리아 Sirtuin
Sirtuins (SIRT)는 NAD+ 양에 의존적으로 표적 단백질의 acetyl기를 때어내는 활성을 가진 효소이다. 포유류에서는 조직의 종류, 세포조직 내 위치, 효소 활성, 표 적 단백질에 따라 Sirt17까지 일곱 가지 종류의 SIRT가 알려져 있으며, 이들 단백 질의 효소 활성을 갖는 중요한 위치는 서로 비슷하다 (Frye, 2000). SIRT는 단백 질 기질에 따라 또는 세포내 위치에 따라 구분 지을 수 있는데 Sirt1, Sirt6, Sirt7 은 핵에 위치하며 Sirt2는 세포질에 위치하고 Sirt3, Sirt4, Sirt5는 미토콘드리아에 위치한다고 알려져 있다. 또 Sirt1, Sirt2, Sirt3, Sirt5, Sirt7은 deacetylase로서의 활성을, Sirt4와 Sirt6은 ADP-ribosyltransferase로써의 활성을 가진다. 각각 SIRT 는 그 위치와 효소 활성에 따라 기능에 맞는 넓은 범위의 단백질을 탈아세틸화 시 킬 수 있다. (Table. 1) (Houtkooper 등, 2012).
Sirt3, Sirt4, Sirt5와 같이 미토콘드리아에 존재하는 SIRT는 ATP 생산, 에너지 대사에 관여하며 세포사멸에도 연관되어있다. 미토콘드리아 SIRT는 100 kDa 크기 의 Sirt1과는 달리 30~40 kDa의 크기로 작으며, N-말단에는 미토콘드리아로 이동 할 수 있도록 하는 염기서열 (Mitochondria targeting sequence)이 구성되어 있다. 미토콘드리아 SIRT는 대사의 변화를 감지하는 역할을 한다. 영양분이 부족하여 세포내 NAD+ 수준이 증가하면 증가된 NAD+ 양에 따라 SIRT의 활성은 증가되는 데, 반면 대사 스트레스가 있는 상황에서는 SIRT의 활성 감소로 인해 지방산 산화, TCA 회로, 산화적 인산화 과정에 필요한 구성체 단백질에 아세틸화가 증가되어 있 음이 보고되었다 (Zhao 등, 2010).
Sirt3은 지방산 산화에 중요한 효소인 long-chain acyl-CoA dehydrogenase (LCAD)의 탈아세틸화를 통해 간에서의 지방의 산화 대사를 조절 한다. Sirt3이 결 손된 마우스에서는 실제로 지방산의 산화가 감소되어 있었고 지방간이 관찰되며 ATP 생산이 감소되어 있음이 보고되었다 (Hirschey 등, 2010). Acetyl-CoA synthetase 2 (AceCS2) 또한 Sirt3에 의해 조절 받는다고 알려져 있으며 AceCS2 가 결손된 마우스에서도 지방산의 산화가 감소되어 있음이 보고되었다 (Hallows 등, 2006). 아마도 Sirt3은 세포내 영양분 부족 스트레스 상황에서 포도당을 제외 한 다른 영양분을 통한 에너지 생산에 중요한 역할을 할 것이라 생각 할 수 있다. 또한 Sirt3은 succinate dehydrogenase (SDH)와 isocitrate dehydrogenase 2 (ICDH2) 그리고 glutamate dehydrogenase (GDH)와 같은 TCA 회로에 관련된 효 소들의 탈아세틸화 조절을 통해 이들 효소 활성화에 관련되어 있다 (Cimen 등, 2010; Schlicker 등, 2008). Sirt3은 전자전달계 구성체 1의 소단위체 NDUFA 9와 succinate dehydrogenase의 상호작용을 통해 이들 단백질을 탈아세틸화 시킴으로 ATP 합성에 영향을 준다고 보고되었다 (Ahn 등, 2008; Cimen 등, 2010).
Sirt4는 TCA 회로에 관련된 효소 glutamate dehydrogenase (GDH)와 상호작용 하며 Sirt3와 달리 Sirt4에 의해서는 GDH의 기능이 ADP-리보실화 활성을 통해 억 제된다. Sirt4가 결손된 마우스에서 GDH의 활성이 증가되어있으며 GDH 활성 증가 에 따라 인슐린 분비도 증가됨이 보고되었다 (Haigis 등, 2006). 또한 Sirt4를 저해
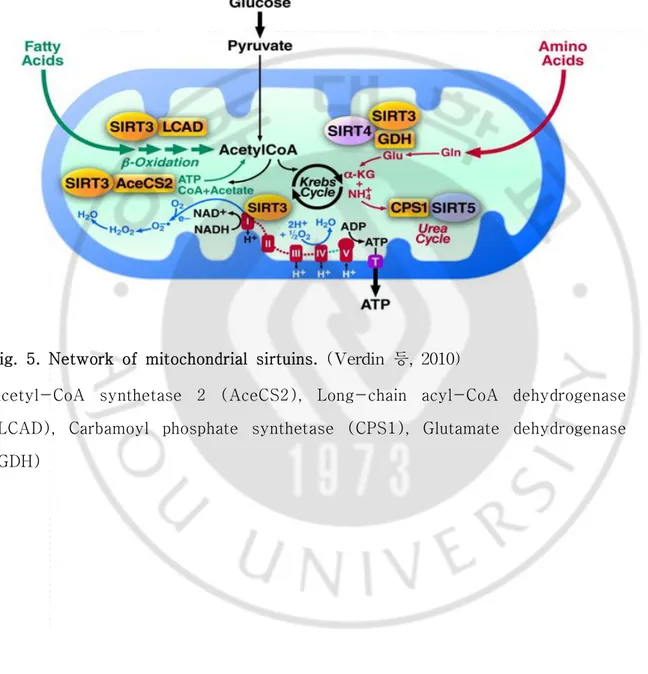
함으로 간이나 지방세포에서 지방산 산화의 증가가 보고되었다 (Nasrin등, 2010). Sirt5는 urea cycle의 첫 번째 속도 조절 효소 (Rate-limiting enzyme)인 carbamoyl phosphate synthetase (CPS1)를 조절한다고 보고되었다 (Nakagawa 등, 2009). 미토콘드리아내 에너지 생산에 관련된 효소와 미토콘드리아 SIRT의 관 계를 그림으로 나타내었다 (Fig. 5).
Fig. 5. Network of mitochondrial sirtuins. (Verdin등,2010)
Acetyl-CoA synthetase 2 (AceCS2), Long-chain acyl-CoA dehydrogenase (LCAD), Carbamoyl phosphate synthetase (CPS1), Glutamate dehydrogenase (GDH)
H. Glutamate dehydrogenase
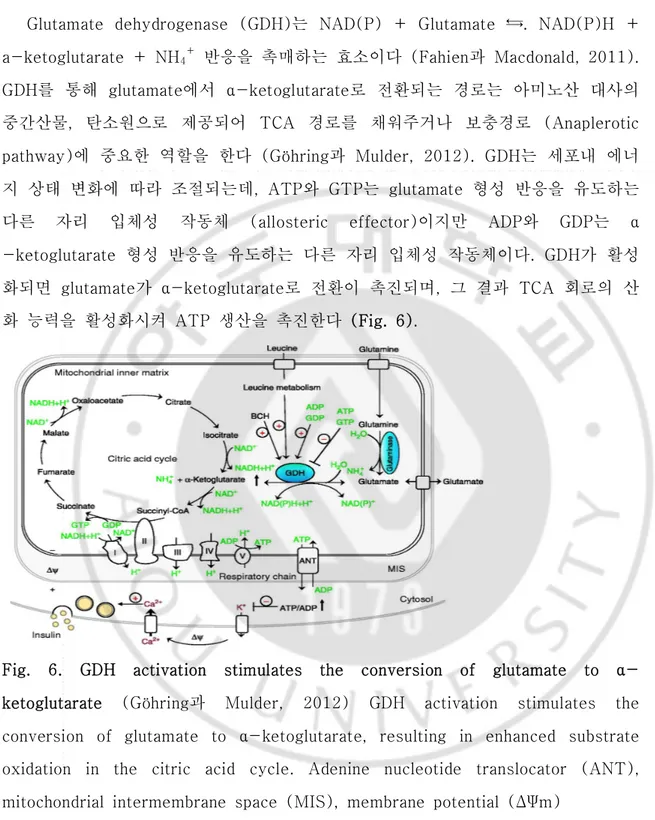
Glutamate dehydrogenase (GDH)는 NAD(P) + Glutamate ⇆. NAD(P)H + a-ketoglutarate + NH4+ 반응을 촉매하는 효소이다 (Fahien과 Macdonald, 2011). GDH를 통해 glutamate에서 α-ketoglutarate로 전환되는 경로는 아미노산 대사의 중간산물, 탄소원으로 제공되어 TCA 경로를 채워주거나 보충경로 (Anaplerotic pathway)에 중요한 역할을 한다 (Göhring과 Mulder, 2012). GDH는 세포내 에너 지 상태 변화에 따라 조절되는데, ATP와 GTP는 glutamate 형성 반응을 유도하는 다른 자리 입체성 작동체 (allosteric effector)이지만 ADP와 GDP는 α -ketoglutarate 형성 반응을 유도하는 다른 자리 입체성 작동체이다. GDH가 활성 화되면 glutamate가 α-ketoglutarate로 전환이 촉진되며, 그 결과 TCA 회로의 산 화 능력을 활성화시켜 ATP 생산을 촉진한다 (Fig. 6).
Fig. 6. GDH activation stimulates the conversion of glutamate to α- ketoglutarate (Göhring과 Mulder, 2012) GDH activation stimulates the
conversion of glutamate to α-ketoglutarate, resulting in enhanced substrate oxidation in the citric acid cycle. Adenine nucleotide translocator (ANT), mitochondrial intermembrane space (MIS), membrane potential (ΔΨm)
베타세포에서 GDH의 활성화제인 루신 (leucin)과 2-Aminobicyclo-(2,2,1)- heptane-2-carboxylic acid (BCH)를 처리하였을 때 GDH의 활성이 증가하며 인슐 린 분비가 촉진되었다 (Panten 등, 1984; Sener 등, 1981). 그러므로 BCH는 베타 세포에서 GDH의 활성을 증가시키는 약제로 생각된다. INS-1 베타세포에 고농도 포도당/지방산을 처리하면 TCA 회로의 중간산물의 부족으로 인해 에너지 생산에 문제가 야기되고 결국 베타세포사멸로 이어지며 이 상황에 BCH를 처리하면 고농도 포도당/지방산 독성에 의한 세포사멸을 억제한다고 보고되었다 (Choi 등, 2011). 이는 BCH 처리를 통한 GDH 활성이 TCA 회로 중간 산물을 증가시킴으로 에너지 생산이 증가되어 고농도 포도당/지방산 독성에 의한 세포사멸을 억제했을 것으로 추측할 수 있다. 또한 db/db 동물 모델에 BCH를 주사 하면 베타세포의 양과 기능을 유지함으로 인슐린 분비를 증가시켜 내당능장애를 개 선하며 항당뇨 효과를 나타내었다 (Han 등, 2012).
I. 연구 목적 고농도 포도당/지방산 독성에 의한 세포사멸 모델에서 NAD+ 전구물질로 사용 되거나, 그 자체로 NAD+를 소비하는 효소의 활성을 저해하는 기능을 가진 NAM 를 처리했을 때 세포사멸이 억제됨을 확인한 바 있다. 그러나 아직 고농도 포도당/ 지방산 독성에 의한 베타세포사멸 모델에서 NAD+ 대사에 대한 이해나 NAM을 통 한 고농도 포도당/지방산에 의한 세포사멸 억제 효과에 대한 정확한 기작이 잘 밝 혀져 있지 않다. 본 연구에서는 고농도 포도당/지방산에 의해 베타세포내 NAD+ 대 사가 어떻게 조절되며 NAD+ 대사가 세포사멸과 어떻게 연관되어 있는지 알아보고 자 하였으며, NAM를 처리했을 때 어떻게 고농도 포도당/지방산 독성에 의한 세포 사멸이 억제되는지 그 기작에 대해 알아보고자 하였다. 1. STZ에 의한 세포사멸 모델과 고농도 포도당/지방산 독성에 의한 세포사멸 모 델에서 NAM을 통한 세포사멸 억제 기작을 비교하였다. 2. 고농도 포도당/지방산 독성에 의한 세포사멸에서 NAM이 항산화효과를 통해 세 포사멸을 억제하는지 조사하였다. 3. 고농도 포도당/지방산 독성에 의한 세포사멸에서 세포내 NAD+ 부족이 세포사 멸을 유도하는데 중요한 역할을 하는지 조사하였다. 4. 고농도 포도당/지방산 독성에 의한 세포사멸에서 NAM의 세포사멸 억제효과가 NAD+ 소비 효소들에 대한 저해 때문인지 알기위해 각 효소들에 대한 siRNA 실험이나 과발현 실험을 통해 고농도 포도당/지방산 독성에서 효소들의 역할 을 조사하였다.
5. 미토콘드리아 SIRT로 알려진 Sirt3, Sirt4가 공동으로 작용하는 표적 단백질인 GDH의 아세틸화 또는 탈아세틸화 활성이 고농도 포도당/지방산 독성에 영향을 미치는지 조사하였다.
Ⅱ. 재료 및 방법
A. 재 료
세포 배양에 사용하는 Fetal bovine serum은 Sigma (Sigma-Aldrich., St. Louis, MO, USA)에서 구입하였고 RPMI 1640은 Cellgro (Mediatech Inc., Manassas, VA, USA)에서 구입하였다. Penicillin과 streptomycin은 Duchefa (Duchefa Biochemie B.V., RV Haarlem, Netherland)에서 구입하였다. 실험에 사용된 약제 glucose, palmitate, streptozotocin (STZ), N-acetylcysteine (NAC), reduced glutathione (GSH), nicotinamide (NAM) β-nicotinamide mononucleotide (NMN), β -nicotinamide adenine dinucleotide (NAD+), L-kynurenine, L-lactate, L-Asparagine, 3-aminobenzamide (3-AB), 5-iodo-6-amino-1,2-
benzopyrone (INH2BP), resveratrol과 sirtinol은 Sigma에서 구입하였다. 3-[4,5-
dimetylthiazol-2-yl]- 2,5-diphenyltetrazoilium bromide (MTT), 과산화수소 (H2O2)는 Duchefa에서 구입하였고, Mito-TEMPOL은 Alexis Biochemicals (Enzo Life Sciences Inc., CA, USA)에서 구입하였고, FK-866은 Cayman Chemical (Cayman Chemical Company., Ann Arbor, MI, USA)에서 구입하였다. IL-1β, TNF-α, and IFN-γ등 사이토카인은 R&D Systems (R&D Systems Inc., Minneapolis, MN, USA)에서 구입하였다. Anti-acetyl-lysine, anti-caspase 3, anti-phospho-Akt, anti-Akt, anti-phospho-eIF2α, anti-eIF2α, anti-CHOP 대 부분의 항체들은 Cell Signaling Technology (Cell Signaling Technology Inc., Beverly, MA, USA)에서 구입하였다. Anti-calnexin 항체는 Stressgen (Enzo Life Sciences Inc., Victoria, BC, Canada)에서 구입하였고 anti-actin과 anti-JNK 항체 는 Santa Cruz (Santa Cruz Biotechnology Inc., Dallas, TX, USA)에서 구입하였고 anti-phospho-JNK 항체는 Invitrogen (Life Technologies Corporation., Carlsbad, CA, USA)에서 구입하였으며 anti-NDUFA9는 Abcam (Abcam plc., Cambridge,
United Kingdom)에서 구입하였다.
B. 방 법
1. 세포주 및 세포 배양
INS-1 베타세포주를 11 mM 포도당이 포함된 RPMI 1640 배지에 10% fetal bovine serum과 100 IU/㎖ penicillin과 100 ㎍/㎖ streptomycin의 항생제를 첨가 하여 5% CO2와 37℃의 온도를 유지하면서 배양하였다.
2. MTT assay
INS-1 세포를 96 well plate에 well당 5×104개씩 분주하고 24시간 동안 배양하 였다. 각 실험 조건에 맞게 약제 처리 후 상등액을 제거하고 MTT (0.5 ㎎/㎖)을 처리 후 배양기에서 한 시간 배양하였다. 상등액을 제거 후 isopropanol 을 첨가하 고 30분간 실온에 둔 다음 570 nm 으로 흡광도를 측정하였다.
3. DNA fragmentation
Cell death detection ELISA Kit (Roche Applied Science., Mannheiem, Germany)를 사용하여 DNA fragmentation 정도를 측정하였다. INS-1 세포를 96 well plate에 well당 5×104개씩 분주하고 24시간 동안 배양하였다. 각 실험 조건에 맞게 약제를 처리한 후 상등액은 제거하고, lysis buffer (Kit 제공) 200 ㎕ 첨가 후 피펫으로 용액을 위아래로 움직여 세포를 용해시킨 후 3000 rpm에 10분간 원
심분리 하였다. 상등액 중 20 ㎕만 취해서 plate (Kit 제공)에 넣은 다음
anti-histon 용액과 anti-DNA PODs 용액을 1/20 농도로 incubation 용액 (Kit 제 공)에 넣은 후 각 well에 80 ㎕씩 넣어 주었다. 상온에서 2시간 반응 후 incubation 용액으로 두 번 씻어 준 후 405 nm로 흡광도를 측정하였다.
INS-1 세포를 6 well plate에 well당 1×106개씩 분주하고 24시간 동안 배양하였 다. 각 실험 조건에 맞게 약제 처리 후 세포들을 수거하여 RIPA buffer (1% triton X-100, 1% sodium deoxycholate, 50 mM NaCl2, 50 mM tris-HCl, 1 mM sodium vanadate, 2 mM PMSF)를 이용하여 세포로부터 단백질을 분리, 정량하여 sample buffer (187 mM tris-HCl pH6.8, 10% SDS, 30% glycerol, 100 mM
DTT, 0.3% bromophenol blue)에 희석하여 5분간 끓인 다음 12%
SDS-polyacrylamid gel에서 전기영동 하였다. Gel에서 PVDF membrane으로 이동 시키고 5% skim milk로 blocking 하였다. 각각의 항체와 반응시키고 horseradish peroxidase가 붙어있는 2차 항체로 반응시켜서 ECL system으로 조사하였다.
5. 세포내 ATP량 측정
ATPLiteTM luminoscence ATP detection assay system (PerkinElmer Life Science., Boston, MA, USA)를 이용하여 세포내 ATP 양을 측정하였다. INS-1 세포를 96 well plate에 well당 5×104개씩 분주하고 24시간 동안 배양하였다. 각 실험 조건별로 처리 후 각각의 well에 세포배양 배지 (100 ㎕)를 제거 하지 않고 lysis buffer (Kit 제공)를 50 ㎕씩 첨가하여 상온에서 700 rpm으로 5분간 흔들어 주었다. lysis buffer을 넣은 각각의 well에 substrate buffer (Kit 제공)를 50 ㎕를 첨가하여 어두운 곳에서 700 rpm으로 5분간 흔들어 주었다. Luminometer로 발광 정도를 측정하였다.
6. 세포내 tNAD량 측정
NAD+/NADH Quantification Kit (BioVision Inc., Mountain View, CA, USA)를 이용하여 세포내 전체 NAD (NAD+, NADH)양을 측정하였다. INS-1 세포를 6 well plate에 well당 1×106개씩 분주하고 24시간 동안 배양하였다. 각 실험 조건별 로 처리된 세포를 수거한 다음, phosphate buffered saline (PBS)로 한번 씻은 후 kit에서 제공하는 assay buffer을 넣고 세포를 용해시켰다. 용해된 세포를 13,000 rpm에 10분간 원심분리 후, 상등액만 분리하여 새로운 튜브에 옮겨 담았다. 옮겨
담은 상등액 중 일부만 (50 ㎕) 만 취하여 plate에 넣은 다음 효소 (50X)를 첨가한 cycling buffer (Kit 제공)를 100 ㎕씩 각 well에 첨가하여 섞어주었다. 5분간 상온 에서 반응시킨 다음 NADH develpoer (Kit 제공)를 처리하여 1시간 동안 상온에서 반응시켰다. 색이 변하면 450 nm에서 흡광도를 측정하였다.
7. 미토콘드리아 분리
QproteomeTM mitochondria isolation kit (Qiagen co., Hilden, Germany)을 사용 하여 미토콘드리아를 분리하였다. INS-1 세포를 100 Φ plate에 1×107개씩 분주하 고 24시간 동안 배양하였다. 실험 조건별로 처리된 세포를 모은 다음 phosphate buffered saline (PBS)로 한번 씻은 후 NAD/NADH extraction buffer (Kit 제공) 을 넣고 세포를 용해시켰다. 용해된 세포를 1000 g에서 10분간 원심분리 후 상등 액은 다른 tube에 모아두고 (미토콘드리아를 제외한 세포구성 물질들을 포함) 침전 물에 다시 disruption buffer (Kit 제공)를 넣고 주사기를 이용해 침전물을 용해시 켰다. 1000 g에서 10분간 원심분리 후 상등액 (미토콘드리아를 포함)을 다시 다른 튜브에 옮겨 담은 후 6000 g에서 10분간 원심분리 하였다. 상등액을 제거 후 침전 물에 storage buffer (Kit 제공)를 넣고 6000 g에서 10분간 원심분리 후 상등액은 제거한다. 침전물 (분리된 미토콘드리아)에 RIPA buffer를 넣고 단백질을 분리하였 다.
8. 형질주입 (Transfection)
NeonTM trangsfection system (Life Technologies Corporation., Carlsbad, CA, USA)을 이용하여 siRNA duplex 또는 Plasmid vector를 INS-1 세포에 형질주입 하였 다. 1×107 INS-1 세포가 담겨있는 튜브에 R buffer (Kit 제공)를 100 ㎕ 씩 넣어주고 siRNA duplex를 pcDNA와 함께 섞은 다음 1650 V, 10 ms width (기계 세팅 조건) 조 건에서 형질주입 하였다. 실험에 사용된 siRNA는 바이오니아 (Daejeon, South Korea) 에서 합성하였으며 siRNA의 염기서열 (Sequence)은 Table 2.에 정리하였다. 실험에 사용된 CMV-Sirt3와 CMV-Sirt4는 한국원자력병원소속의 이기호 박사님이 제공해
주셨으며 pc-Visfatin은 그린진 바이오텍 (GreenGene Biotech., Gyouggido, South Korea)에서 제작하였다. Pc-CD38, pc-NADK, pc-GDH ADPr, pc-GDH KR, pc-GDH KQ DNA vector는 실험실에서 제작하였다.
9. RT-PCR
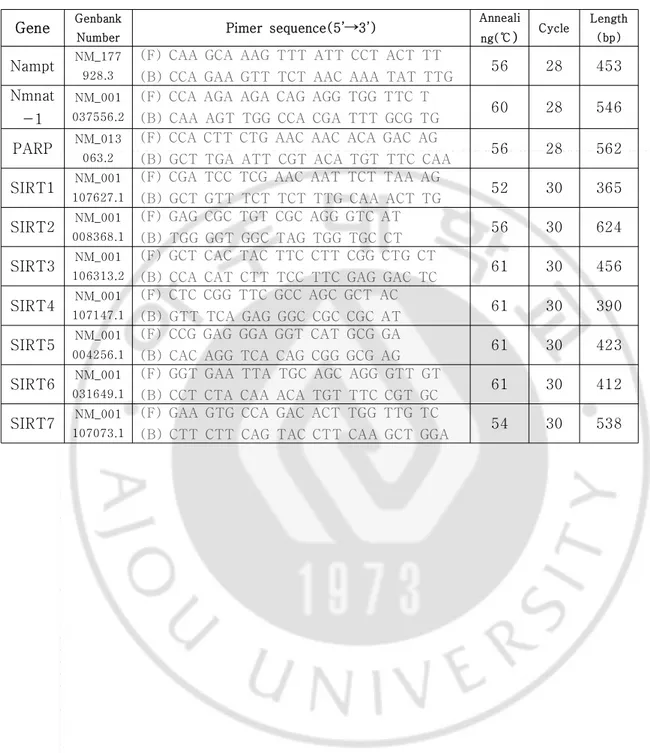
수거한 세포를 PBS로 2회 씻어준 후, RNA iso plus (TAKARA biotechnology co., Dalian,Japan)를 이용해 전체 RNA를 뽑고 정량하였다. 1㎍의 RNA를 1000 U AMV 0.5 ㎕, 2.5 mM dNTP 4 ㎕, Random 9mer 1 ㎕, RNase inhibitor 0.5 ㎕, Mgcl2 4 ㎕, 10X RT buffer를 이용해서 30℃ 10분, 42℃ 30분, 99℃ 5분 동안 역전사 반응을 시켰 다. 얻어진 cDNA로 PCR 하였다. 실험에 사용된 프라이머 (primer)는 Table 3.에 정 리하였다. 1.2% agarose gel에서 PCR 반응물을 전기영동 후 EtBr로 염색하여 관찰 하였다.
10. Plasmid vector 구축 가. pcCD38
백서 소도세포를 주형으로 얻은 DNA로 (F) 5'-CAG GGT ACC AAT GGC CAA CTA TGA ATT TTC C (B) 5'-CAG CTC GAG TCC TTC ACA CAT TAA GTC TAC AT 프라이머를 이용하여 약 0.92 kbp CD38 DNA를 증폭시켰다. 증폭 시킨 CD38 DNA와 pcDNA3를 제한효소 XhoⅠ와 KpnⅠ으로 절단 후 두 유전자를 접합 (ligation)하여 pcCD38 plasmid vector를 구축하였다. 제작한 palsmid vector 의 염기서열은 마크로젠 (Seoul, Korea)에서 분석하였다.
나. pcNADK
INS-1 베타세포를 주형으로 얻는 DNA로 (F) 5'-CAG AAG CTT CAT ATC AAC TAT GGA AAT GGA ACA A (B) 5'-CAG CTC GAG AAG CTG GGA AAT GTA GTC TAG C 프라이머를 이용하여 약 1.4 kbp NADK DNA를 증폭시 켰다. 증폭시킨 NADK와 pcDNA3를 제한효소 Hind Ⅲ와 Xho Ⅰ으로 절단 후 두 유전자를 접합하여 pcNADK plasmid vector를 구축하였다. 제작한 palsmid vector
의 염기서열은 마크로젠에서 분석하였다.
11. Glutamate dehydrogenase mutant plasmid vector 구축 가. ADP-ribosylation site mutant (C172A)
INS-1 베타세포를 주형으로 얻은 DNA를 (F) 5'-GAC GGT ACC ATG TAC CGC CGT CTG GGC GAA G (B) 5'-GGC ACA TCA ACC ACT GCA GCC TTG TAG GTC ATT AGG GA 프라이머로 PCR 생성물 1을 만들고 (F) 5'-TCC CTA ATG ACC TAC AAG GCT GCA GTG GTT GA T GTG CC (B) 5‘-GAC CTC GAG TCT ATG TGA AGG TCA CGC CAG C 프라이머로 PCR 생성물 2를 만들 었다. 두 개의 PCR 생성물을 섞은 다음 이 유전자를 주형으로 하여 (F) 5'-GAC GGT ACC ATG TAC CGC CGT CTG GGC GAA G (B) 5‘-GAC CTC GAG TCT ATG TGA AGG TCA CGC CAG C 프라이머로 DNA를 증폭시켜 mutant DNA를 생성하였다. 만들어진 PCR 생성물과 pcDNA3를 제한효소 Kpn Ⅰ과 Xho Ⅰ으로 절단 후 접합하여 GDH-ADP 리보실화 site mutant plasmid DNA를 제작하 였다. 제작한 palsmid vector의 염기서열은 마크로젠에서 분석하였다.
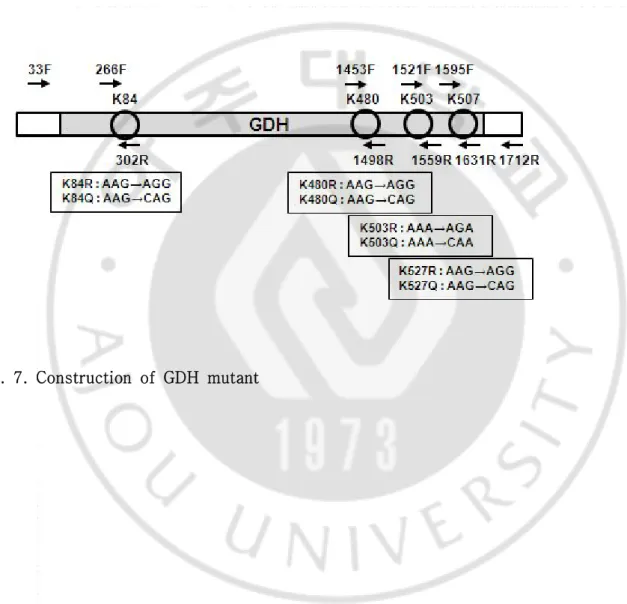
나. GDH KR, KQ mutant
GDH의 아미노산중 리신 (Lysine : K)이 아르지닌 (Arginine : R)으로 치환되거 나 리신 (Lysine : K)이 글루타민 (Glutamin : Q)으로 치환된 변형된 형태의 GDH DNA를 얻기 위해 (Fig. 7)과 같이 프라이머를 디자인하였다. INS-1 베타세포를 주형으로 하여 DNA를 얻은 다음 Table 4.에 적혀있는 프라이머로 각각의 서열에 변형된 DNA를 증폭하였다. 각각의 변형된 PCR 생성물을 주형으로 4군데 모두 변 형시키고자하는 염기서열로 변환된 2차 PCR 생성물을 증폭시켰다. 변형된 GDH DNA와 pcDNA3를 제한효소 Kpn Ⅰ과 Xho Ⅰ으로 절단 후 접합하여 GDH- 아세 틸화 site mutant plasmid DNA를 제작하였다. 제작한 palsmid vector의 염기서열 은 마크로젠에서 분석하였다.
12. 통계
3번 이상의 독립적인 실험으로 통계 처리하였으며 오차는 SE값으로 나타내었다. 통 계 값은 Student's t test를 이용하여 구하였으며 군간 p 값이 0.05이상일 때 유의 성이 있다고 표시하였다.
Genes Genbank Number siRNA sequence (5’→3’) GFP GU983383 (S) GUU CAG CGU GUC CGG CGA G
(A) CUC GCC GGA CAC GCU GAA C Nampt NM_177928.3
(S) CUG UUC ACA GUG GAA AAC A (A) UGU UUU CCA CUG UGA ACA G
Nmnat1 NM_001037556.2
(S) GAC UUG CCA UCC UGU UUU A (A) UAA AAC AGG AUG GCA AGU C
CD38 D29646.1
(S) CCA CAU UGG AGU GAA AAU UUU (A) AAU UUU CAC UCC AAU GUG GUU
NADK BC090019.1
(S) CAA AGA GUG UGC UUG UUA UUU (A) AUA ACA AGC ACA CUC UUU GUU
PARP NM_013063.2 (S) GAA AAG AGG AUG AAA CUA A
(A) UUA GUU UCA UCC UCU UUU C
SIRT1 NM_001107627.1 (S) GAA AAG ACC CAA GAC CAU U
(A) AAU GGU CUU GGG UCU UUU C
SIRT2 NM_001008368.1 (S) CAG UUC AAG CCG ACC AUC U
(A) AGA UGG UCG GCU UGA ACU G
SIRT3 NM_001106313.2 (S) UUU GAA CUA GGC UUC UUU
(A) AAA GAA GCC UAG UUC AAA
SIRT4 NM_001107147.1
(S) CCA GCG UGU GAA AGA AGC U (A) AGC UUC UUU CAC ACG CUG G
SIRT5 NM_001004256.1
(S) CGU GUG GCA AUG UUC CUG A (A) UCA GCA ACA UUG CCA CAC G
SIRT6 NM_001031649.1 (S) GUG UUC AGC ACA GUU CCU U
(A)AAG GAA CUG UGC UGA ACA C
SIRT7 NM_001107073.1 (S) GUC CGA AGU GCC AGA CAC U
(A) AGU GUC UGG CAC UUC GGA C Table 2. Genbank number and nucleotide sequence of siRNAs