Hereditary
Hereditary
Hemolytic
Hemolytic
Anemia
Anemia
Young-shil, Park, M.D.
Young-shil, Park, M.D.
Division of Hematology-Oncology Department of PediatricsIntroduction
Hemolysis
the premature destruction of red blood cells (RBCs)
Normal RBC
survival time ; 110–120 days (half-life, 55–60 days) approximately 0.85% of RBCs (the senescent ones)
; removed each day and replaced by the marrow to maintain the RBC count
During hemolysis
RBC survival is shortened the RBC count falls
erythropoietin is increased
the stimulation of marrow activity results in heightened
RBC production
an increased percentage of reticulocytes in the
The marrow can increase
its output two- to threefold acutely, with a maximum of six- to eightfold
reticulocyte count
hemolysis
response to acute blood loss
for a short period after replacement therapy for iron,
Figure. Red cell destruction and catabolism of hemoglobin (Hb) based on the description by Hillman and Finch. Fe, iron. (From Hillman RS, Finch CA: Red Cell Manual. Philadelphia, FA Davis, 1983.)
Hereditary spherocytosis
Hereditary elliptocytosis
Hereditary stomatocytosis
Hemoglobin disorders
Enzyme defects
common cause of hemolysis and hemolytic
anemia
prevalence of approximately 1/5,000 in people of
Northern European descent
most common inherited abnormality of the red
blood cell (RBC) membrane.
without anemia
with minimal hemolysis
Etiology
autosomal dominant
less frequently, autosomal recessive
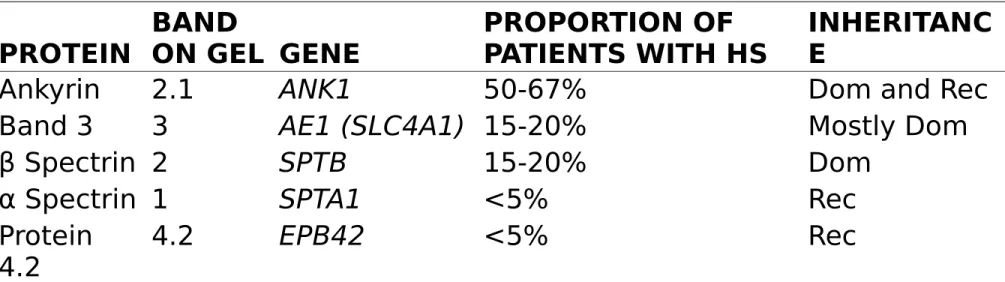
most common molecular defects
Table 458-1. Common Gene Mutations in Hereditary Spherocytosis (HS) PROTEIN BAND ON GEL GENE PROPORTION OF PATIENTS WITH HS INHERITANC E
Ankyrin 2.1 ANK1 50-67% Dom and Rec
Band 3 3 AE1 (SLC4A1) 15-20% Mostly Dom
β Spectrin 2 SPTB 15-20% Dom
α Spectrin 1 SPTA1 <5% Rec
Protein
loss of membrane surface area without a
proportional loss of cell volume → sphering of the RBCs
associated increase in cation permeability cation transport
decreased deformability of the spherocytic RBCs
impairs cell passage from the splenic cords to the splenic sinuses
spherocytic RBCs are destroyed prematurely in
the spleen.
Splenectomy markedly improves RBC life span
Clinical manifestations
in the newborn
anemia and hyperbilirubinemia
⇒ phototherapy or exchange transfusions
The severity in infants and children is
After infancy
splenomegaly
pigmentary (bilirubin) gallstones ; as early as age 4–5 yr
susceptible to aplastic crisis
primarily as a result of parvovirus infection Such erythroid marrow failure
⇒ profound anemia (hematocrit <10%) high-output heart failure, hypoxia cardiovascular collapse, and death
Laboratory findings
reticulocytosis & indirect
hyperbilirubinemia
Peripheral blood smear
Hereditary Spherocytosis
confirmed with an
osmotic fragility test
RBC membrane protein analysis using
gelDifferential diagnosis
large numbers of spherocytes
1) Isoimmune hemolytic disease
, particularly due to ABO incompatibility
2) thermal injury
3) clostridial septicemia with exotoxemia 4) Wilson disease
Treatment
splenectomy
eliminates most of the hemolysis
After splenectomy, osmotic fragility often
improves
because of diminished splenic conditioning and less
RBC membrane loss; the anemia, reticulocytosis, and hyperbilirubinemia then resolve.
For patients with more severe anemia
reticulocytosis
those with hypoplastic or aplastic crises
poor growth cardiomegaly splenectomy
-> recommended after age 5-6 yr to avoid the heightened risk of postsplenectomy sepsis in younger children
Vaccines (conjugated and/or capsular) for
encapsulated organisms, such as
pneumococcus, meningococcus, and
Haemophilus influenzae type b
,
should be administered before splenectomy
prophylactic oral penicillin V (age <5 yr, 125 mg
twice daily; age 5 yr through adulthood, 250 mg
twice daily) administered thereafter.
Hereditary Elliptocytosis
less common disorder than spherocytosis that also
varies markedly in severity
The blood film is the most important test to establish
hereditary elliptocytosis
Increased thermal instability
no treatment is necessary
Splenectomy
Hereditary stomatocytosis
Hemoglobinopathies
Hemoglobin ; tetramer consisting of 2 pairs of
globin chains.
Abnormalities in these proteins are referred to
Hb F (α2γ2) Hb A2 (α2δ2)
Sickle Cell Disease
Hemoglobin S (Hb S)
result of a single base pair change, thymine for
adenine, at the 6th codon of the β-globin gene
In sickle cell anemia, Hb S ; commonly as high as
90% of the total hemoglobin.
Hb SS
In sickle cell disease, Hb S is >50% of all
Treatment
need continuous treatment
Supplementation with folic acid, an essential element
in producing red blood cells,
Children with sickle cell anemia ; deficient levels of
serum opsonins of the alternate complement pathway against pneumococci.
increased risk for infection and death as a result of
bacterial infection, particularly with encapsulated
organisms, such as Streptococcus pneumoniae and Haemophilus influenzae type B
should receive prophylactic oral penicillin VK at least
Thalassemia Syndromes
genetic disorders in globin chain production
β-thalassemia ; either a complete absence of β-globin gene
production (β0-thalassemia) or a partial reduction (β+
-thalassemia)
α-thalassemia ; α-globin gene production is either absent or
partially reduced
The primary pathology in thalassemia stems from the quantity of
globin gene production, whereas in sickle cell disease, the primary pathology is the quality of globin production.
particularly prevalent among Mediterranean people Middle Easterners, North Africans, and South Asians
Figure 462-7 Ineffective
erythropoiesis in an
untransfused 3 yr old patient with thalassemia major. A,
Massive widening of the diploic spaces of the skull as seen on MRI. B, Radiographic
appearance of the trabeculae as seen on plain radiograph, and C, obliteration of the maxillary
sinuses with hematopoietic tissue as seen on CT scan.
chronic blood transfusion therapy
iron chelation
Splenectomy
Enzyme defects
Various red blood cell (RBC) enzymatic defects
produce hemolytic anemias.
1. Pyruvate Kinase Deficiency
The clinical manifestations vary from severe neonatal hemolytic
anemia to mild, well-compensated hemolysis first noted in adulthood.
Severe jaundice and anemia in the neonatal period kernicterus
The hemolysis in older children and adults varies in severity, with
hemoglobin values ranging from 8-12 g/dL associated with some pallor, jaundice, and splenomegaly.
2. Glucose-6-Phosphate Dehydrogenase
Deficiencies
episodic hemolytic anemia induced by infections, certain drugs or, rarely, fava beans, and spontaneous chronic nonspherocytic hemolytic anemia
In the usual pattern of G6PD deficiency, symptoms develop
24-48 hr after a patient has ingested a substance that has oxidant properties.
Table 463-1. Agents Precipitating Hemolysis in Glucose-6-Phosphate Dehydrogenase Deficiency MEDICATIONS Antibacterials Sulfonamides Trimethoprim-sulfamethoxazole Nalidixic acid Chloramphenicol Nitrofurantoin Antimalarials Primaquine Pamaquine Chloroquine Quinacrine Others Phenacetin Vitamin K analogs Methylene blue Probenecid Acetylsalicylic acid Phenazopyridine CHEMICALS Phenylhydrazine Benzene Naphthalene ILLNESS Diabetic acidosis Hepatitis Sepsis