185 책임저자:박옥진, 305-811, 대전시 유성구 전민동 461-6
한남대학교 대덕 밸리 캠퍼스 식품영양학과 Tel: 042-629-8793, Fax: 042-629-8789 E-mail: [email protected]
접수일: 2010년 4월 1일, 1차수정일: 2010년 7월 7일, 2차수정일: 2010년 7월 9일, 게재승인일: 2010년 7월 13일
Correspondence to:Ock Jin Park
Department of Food and Nutrition, Hannam University Daedeok Valley Campus, 461-6, Jeonmin-dong, Yuseong-gu, Daejeon 305-811, Korea Tel: +82-42-629-8793, Fax: +82-42-629-8789
E-mail: [email protected]
Effects of Celecoxib and Compound C on AMPKα1/Akt Signaling in MCF-7 Breast Cancer Cells
Yun-Kyoung Lee1, Song Yi Park2, Sol Hwa Lee2, Young Min Kim2 and Ock Jin Park1 Departments of 1Food and Nutrition, 2Biological Sciences,
Hannam University Daedeok Valley Campus, Daejeon 305-811, Korea
Most breast cancers have characteristics of over-activated Akt, a major stimulator of cell growth and tumor progression. Thus, suppressing Akt activity can provide an effective way for tumor control. In this study, we have focused on the regulatory effects of AMPKα1 and cyclooxygenase-2 (COX-2) on cell survival signal in MCF-7 breast cancer cells using their specific inhibitors, Compound C and Celecoxib for AMPKα1 and COX-2, respectively. Our results showed that the inhibition of AMPKα1 by Compound C increased both Akt and COX-2 levels. Blocking COX-2 expression by Celecoxib increased AMPKα1, and surprisingly it increased Akt in MCF-7 cells. To determine the relationship between Akt and COX-2 signals, we used the cox-2 negative and positive cells. In cox-2 positive cells, Celecoxib decreased COX-2 level as well as Akt activity, but activated AMPKα1. In addition, treatment of Celecoxib and LY294002, Akt inhibitor reduced cell growth of MCF-7 cells. Therefore, these results suggest that there might be existence in mutual regulatory interactions between COX-2 and Akt signals in only cancerous applicable cells. Moreover, AMPKα1 can act as a negative regulator of Akt and COX-2 signals, and targeting AMPKα1 to control tumor growth can be an effective way due to its inhibitory effects of Akt against breast cancers. (Cancer Prev Res 15, 185-189, 2010)
Key Words: Akt, AMP-activated protein kinase, Celecoxib, Compound C, Cyclooxygenase-2
INTRODUCTION
Over-phosphorylated Akt is commonly observed in breast cancers and blocking Akt-regulated signal pathways has been emerged as an effective strategy for cancer therapy because of its involvement in the inhibition of apoptosis and the activation of survival and metastatic pathways.1∼3) Akt, a serine/threonine protein kinase is phosphorylated by phosphoinositide-3-kinase (PI3K) in response to growth factors such as insulin like growth factor-1 (IGF-1) and epidermal growth factor (EGF) or other mitogenic signals.4,5) Once activated, Akt stimulates phosphorylation of tuberous sclerosis protein 2 (TSC2), which activates mammalian target of rapamycin (mTOR), a major
controller of protein synthesis related to cell survival factors such as nutrient transporters, angiogenic factors and cell cycle controllers.6∼8) Thus, the over-activation of Akt permits cell to continuously proliferate, survive and metastasize. In addition, Akt is also activated by inflammatory signals such as pro- inflammatory cytokines. Phosphorylated Akt stimulates inflam- matory signals through activating several mitogen-activated protein kinase (MAPK) such as ERK and p38.9,10) It has been reported that Akt negatively regulates glycogen synthase kinase 3-β (GSK3β), which increases the transcriptional activity of NF-kB p65 in human monocytes.11) These findings suggest that the inhibition of Akt phosphorylation by both of growth factor and inflammatory signals can provide more effective ways for controlling cancers.
Fig. 1. AMPKα1 is involved in growth inhibition of MCF-7 breast cancer cells. Cells were treated with 20μM of LY294002, 10μM of Compound C and 50μM of Celecoxib for 6 h and cell viability was measured by MTT assay. Values with different superscript represent significantly different at a,bp
<0.05 (each experiment’s n=3).
AMPK, an energy sensing protein kinase, has been reported to suppress Akt through the activation of phosphatase such as phosphatase and tensin homolog (PTEN) or protein phosphat- ase 2A (PP2A). PTEN has been known to inhibit PI3K phosphorylation induced by IGF-1 and PP2A has been reported to dephosphorylate Akt, directly.12,13) It also has been demonstrated that AMPK inhibition by Compound C, synthetic inhibitor of AMPK, or siRNA of AMPK increased Akt phosphorylation levels in cancer cells.14) Although the exact molecular mechanisms of AMPK to inhibit Akt are not fully elucidated, the possibility of AMPK as a negative regulator of Akt has been suggested. Therefore, in this study, we have investigated the effects of Compound C, and Celecoxib, a selective COX-2 inhibitor and anti-inflammatory drug, on Akt activity in MCF-7 breast cancer cells as well as cox-2 negative and positive cells. We demonstrated that AMPK α1 activation is necessary for Akt inhibition, however inhibiting inflammatory responses did not appear to be responsible for Akt suppression.
MATERIALS AND METHODS 1. Cell culture and reagents
MCF-7 cells were obtained from the American Type Culture Collection (Manassas, VA, USA) and grown in RPMI 1640 medium containing 10% fetal bovine serum (Gibco, USA) at 37oC in a 5% CO2 atmosphere. Compound C was obtained from Calbiochem (San Diego, CA, USA) and LY294002 was purchased from Tocris (Bristol, UK). Celecoxib (Celebrex) was supplied from Pharmacia (Seoul, Korea). Specific antibodies that recognize the phosphorylated forms of AMPKα1Thr172, ACC and Akt were obtained from Cell Signaling Technology (Danvers, MA). Anti-COX-2 was purchased from Santa Cruz Biotechnology (San Diego, CA, USA) and anti-β-actin was obtained from Sigma.
2. Measurement of cell viability
Cells seeded on 96-well micro plates at 4,000 cells/well were incubated with CoCl2 or curcumin for indicated concentrations or time periods. Respective medium was removed, and the cells were then incubated with 100μl of MTT solution (2 mg/ml MTT in PBS) for 4 h. Converted purple formazan dye from MTT was solubilized in DMSO, and optical densities were measured at 560 nm.
3. Western blot analysis
The cells were washed with phosphate-buffered saline (PBS), scraped into RIPA lysis buffer (50 mM Tris-HCl [pH 8.0], 1%
NP-40, 0.5% sodium deoxycholate, 150 mM NaCl, 1 mM PMSF) with phosphatase inhibitors (Sigma) and subjected to western blot analysis with specific antibodies. Then, the proteins were visualized by enhanced chemiluminescence (Intron, Korea) and detected using the LAS4000 chemilumines- cence detection system (Fuji, Tokyo, Japan).
4. Statistical analysis
Cell viability were statistically analyzed using unpaired t-test (SPSS, Chicago, IL). p<0.05 was considered statistically significant.
RESULTS
1. Inhibition of AMPKα1 by Compound C increases Akt activity and COX-2 expression in MCF-7 breast cancer cells
To examine whether AMPKα1 inhibition or COX-2 inhibition regulate Akt activity, we treated specific inhibitors of Akt, AMPKα1 and COX-2 with indicated concentrations and observed changes of molecular levels. Our results showed that AMPKα1 inhibition by Compound C increased
Fig. 2. AMPKα1 inhibition increased phospho-Akt and COX-2 expressions in MCF-7 breast cancer cells. Cells were treated with 20μM of LY294002, 10μM of Compound C and 50μM of Celecoxib for 6 h and total proteins were subjected to Western blot analysis using phospho-ACC, phospho-AMPKα 1, phospho-Akt, COX-2 and β-actin (loading control) anti- bodies.
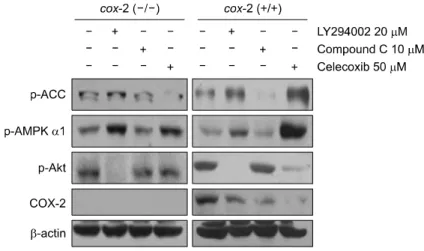
Fig. 3. Blocking inflammatory responses by Celec- oxib decreased Akt activity in cox-2 positive cells.
Cox-2 negative and positive cells were treated with 20μM of LY294002, 10 μM of Compound C and 50 μM of Celecoxib for 6 h and total proteins were subjected to Western blot analysis using phospho- ACC, phospho-AMPKα1, phospho-Akt, COX-2 and β-actin (loading control) antibodies.
phospho-Akt level and COX-2 expression and stimulated cell growth (Fig. 1, 2). It implies that AMPKα1 acts as a negative regulator of Akt as well as COX-2 and it is supported by the up-regulation of AMPKα1 following treatment of LY294002 and Celecoxib. In addition, blocking inflammatory responses by Celecoxib decreased COX-2 expression but increased phospho- Akt levels although Akt inhibition by LY294002 suppressed COX-2 expression. The observed activation of p-ACC (The best-characterized phosphorylation site of AMPK, hence the phosphorylation level of acetyl-CoA carboxylase serine 79) expression by the treatment with COX-2 inhibitor, Celecoxib presents AMPK activation was enhanced by inhibiting COX-2.
These results suggest that the inflammatory stimuli have little
effect on Akt phosphorylation, and Akt may partially regulate COX-2 expressions in MCF-7 breast cancer cells. Additionally both of LY294002 and Celecoxib showed slight growth inhibitory effects of MCF-7 breast cancer cells.
2. Celecoxib treatment decreased Akt levels in cox-2 positive cells
To determine the effect of Celecoxib on Akt activity, we used cox-2 negative and positive cells. In similar to results in MCF-7 cells, Compound C treatment increased Akt levels in both of cox-2 negative and positive cells. Also, LY294002 treatment increased AMPKα1 activity and decreased COX-2 expression. However, Celecoxib showed different patterns with MCF-7 cells. It decreased COX-2 level as well as Akt activity in cox-2 positive cells although it was not observed in cox-2 negative cells. Additionally, Celecoxib strongly activated AMPKα1 in both of cells (Fig. 3). These results suggest that blocking inflammatory responses could be partially involved in Akt suppression in normal cells and AMPKα1 activation is major inhibitory pathway for Akt in both of normal and cancerous cells.
DISCUSSION
In this study, we investigated the possibilities of AMPKα1 and COX-2 signals as a negative or positive regulator of Akt signal in cancer or normal cells using specific inhibitors, Compound C and Celecoxib, respectively. Akt has been known a promising target molecule in cancer therapy due to its stimulation of cell proliferation and survival pathways such as angiogenesis and metastasis. We have demonstrated that
AMPKα1 acts as a negative regulatory molecule of Akt, and COX-2 may not be crucial element of Akt regulation in MCF-7 breast cancer cells.
AMPK has been reported to inhibit abnormal cell proliferation and activate apoptotic pathway in several cancer cells.15∼17) Additionally one possible explanation of these AMPKα1 actions is its regulatory effects of Akt activity. It has been reported that AMPK induced apoptosis through the blockage of Akt-stimulated survivin expression, a putative IAP, in lung cancers.14) Our results showed that the AMPKα1 inhibition by Compound C stimulated cell growth and increased Akt level. The Akt ingibition by LY294002 also decreased cell viability but increased AMPKα1 activity. It implies that AMPKα1 controls cancer cell growth through inhibiting Akt signals in MCF-7 breast cancer cells. However, the effect of AMPKα1 on regulation of cell viability seems not to be dependent on Akt inhibition only, because AMPKα 1 inhibition also increased COX-2 expression. We previously reported that activated AMPKα1 by selenium induced apoptosis through suppressing ERK-COX-2 pathway in colon cancer cells.18) Our results also showed AMPKα1 should act as a negative regulator at upstream signal of COX-2 in both of cancerous cells and normal cells. It seems evident that activating AMPKα1 could provide beneficial effects for cancer control through inhibiting Akt and COX-2 signals, possible downstream target molecules.
Further, we next examined whether the inhibition of inflammatory responses could suppress Akt phosphorylation, because inflammatory stimuli such as LPS, TNF, and IL-6 have reported to increase Akt activity.19,20) Our results showed that blocking inflammatory responses could not regulate Akt activity, and AMPKα1 was ineffective in inhibiting Akt during this process. Although Celecoxib, an anti-inflammatory drug, exerted its anti-cancer effects through inhibiting cell growth and COX-2 expression, it increased p-Akt levels in MCF-7 breast cancer cells. In cox-2 positive cells, Celecoxib showed strong inhibitory effects on Akt signals as well as COX-2. Therefore, it seems that the regulatory effect of Celecoxib is dependent on cell types and there might be existed mutual regulatory interactions between COX-2 and Akt signals in only cancerous applicable cells.
In conclusion, we observed that activating AMPKα1 is an important factor for inhibition of Akt as well as COX-2 signals and suppressing COX-2 signals could not fully regulate Akt
activity in MCF-7 breast cancer cells. Therefore, our findings suggest that targeting AMPKα1 activation appears to be an attractive way for controlling cancer growth or apoptosis against breast cancers, possibly regulating both Akt and COX-2 via the activation of AMPKα1, whereas inhibiting only COX-2 by a pharmacological agent may not be an effective way to control Akt as well as AMPKα1.
CONCLUSION
The inhibition of AMPK by Compound C elevated both Akt and COX-2 levels in cancerous MCF-7 cells, and the suppression of COX-2 with a pharmacological COX-2 inhibitor increased Akt even though AMPK was greatly elevated by blocking COX-2. Therefore, AMPK inhibitor is an effective controller of Akt or COX-2 while COX-2 inhibitor may not be the major controller of Akt and AMPK.
ACKNOWLEDGEMENT
This work was supported by Hannam University Fund 2010.
REFERENCES
1) Liu W, Bagaitkar J, Watabe K. Roles of AKT signal in breast cancer. Front Biosci 12, 4011-4019, 2007.
2) Cheng GZ, Park S, Shu S, He L, Kong W, Zhang W, Yuan Z, Wang LH, Cheng JQ. Advances of AKT pathway in human oncogenesis and as a target for anti-cancer drug discovery. Curr Cancer Drug Targets 8, 2-6, 2008.
3) Cicenas J. The potential role of Akt phosphorylation in human cancers. Int J Biol Markers 23, 1-9, 2008.
4) Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov 8, 627-644, 2009.
5) Carnero A, Blanco-Aparicio C, Renner O, Link W, Leal JF.
The PTEN/PI3K/AKT signalling pathway in cancer, thera- peutic implications. Curr Cancer Drug Target 8, 187-198, 2008.
6) Memmott RM, Dennis PA. Akt-dependent and -independent mechanisms of mTOR regulation in cancer. Cell Signal 21, 656-664, 2009.
7) Huang J, Manning BD. A complex interplay between Akt, TSC2 and the two mTOR complexes. Biochem Soc Trans 37, 217-222, 2009.
8) Pérez-Tenorio G, Stål O. Activation of AKT/PKB in breast cancer predicts a worse outcome among endocrine treated patients. Br J Cancer 86, 540-545, 2002.
9) Cakir M, Grossman AB. Targeting MAPK (Ras/ERK) and
PI3K/Akt pathways in pituitary tumorigenesis. Expert Opin Ther Targets 13, 1121-1134, 2009.
10) Albrecht DS, Clubbs EA, Ferruzzi M, Bomser JA. Epigallo- catechin-3-gallate (EGCG) inhibits PC-3 prostate cancer cell proliferation via MEK-independent ERK1/2 activation. Chem Biol Interact 171, 89-95, 2008.
11) Guha M, Mackman N. The phosphatidylinositol 3-kinase-Akt pathway limits lipopolysaccharide activation of signaling pathways and expression of inflammatory mediators in human monocytic cells. J Biol Chem 277, 32124-32132, 2002.
12) Huang X, Wullschleger S, Shpiro N, McGuire VA, Sakamoto K, Woods YL, McBurnie W, Fleming S, Alessi DR. Impor- tant role of the LKB1-AMPK pathway in suppressing tumori- genesis in PTEN-deficient mice. Biochem J 412, 211-221, 2008.
13) Kim KY, Baek A, Hwang JE, Choi YA, Jeong J, Lee MS, Cho DH, Lim JS, Kim KI, Yang Y. Adiponectin-activated AMPK stimulates dephosphorylation of AKT through protein phosphatase 2A activation. Cancer Res 69, 4018-4026, 2009.
14) Jin Q, Feng L, Behrens C, Bekele BN, Wistuba II, Hong WK, Lee HY. Implication of AMP-activated protein kinase and Akt-regulated survivin in lung cancer chemopreventive activities of deguelin. Cancer Res 67, 11630-11639, 2007.
15) Hsu YC, Meng X, Ou L, Ip MM. Activation of the AMP- activated protein kinase-p38 MAP kinase pathway mediates apoptosis induced by conjugated linoleic acid in p53-mutant mouse mammary tumor cells. Cell Signal 22, 590-599, 2010.
16) Park HU, Suy S, Danner M, Dailey V, Zhang Y, Li H, Hyduke DR, Collins BT, Gagnon G, Kallakury B, Kumar D, Brown ML, Fornace A, Dritschilo A, Collins SP. AMP- activated protein kinase promotes human prostate cancer cell growth and survival. Mol Cancer Ther 8, 733-741, 2009.
17) Rattan R, Giri S, Singh AK, Singh I. 5-Aminoimidazole-4- carboxamide-1-beta-D-ribofuranoside inhibits cancer cell proli- feration in vitro and in vivo via AMP-activated protein kinase.
J Biol Chem 280, 39582-39593, 2005.
18) Hwang JT, Kim YM, Surh YJ, Baik HW, Lee SK, Ha J, Park OJ. Selenium regulates cyclooxygenase-2 and extra- cellular signal-regulated kinase signaling pathways by activat- ing AMP-activated protein kinase in colon cancer cells. Cancer Res 66, 10057-10063, 2006.
19) Shishodia S, Koul D, Aggarwal BB. Cyclooxygenase (COX)-2 inhibitor celecoxib abrogates TNF-induced NF-kappa B activation through inhibition of activation of I kappa B alpha kinase and Akt in human non-small cell lung carcinoma:
correlation with suppression of COX-2 synthesis. J Immunol 173, 2011-2022, 2004.
20) Dagia NM, Agarwal G, Kamath DV, Chetrapal-Kunwar A, Gupte RD, Jadhav MG, Dadarkar SS, Trivedi J, Kulkarni- Almeida AA, Kharas F, Fonseca LC, Kumar S, Bhonde MR.
A preferential p110{alpha}/{gamma} PI3K inhibitor attenu- ates experimental inflammation by suppressing the production of proinflammatory mediators in a NF-{kappa}B-dependent manner. Am J Physiol Cell Physiol 298, 929-941, 2010.