Copyrightⓒ 2011, The Korean Academy of Oral Biology
13
Journal of Oral Biology
A Natural Product, Chios Gum Mastic, Induces the Death of HL-60 Cells via Apoptosis and Cell Cycle Arrest
Byung-Chan Koo, Duck-Han Kim, In-Ryoung Kim,Gyoo-Cheon Kim, Hyun-Ho Kwak, and Bong-Soo Park*
Department of Oral Anatomy, School of Dentistry, Pusan National University (received December 20, 2010 ; revised February 14, 2011 ; accepted March 4, 2011)
Chios gum mastic (CGM) is produced from Pistiacia lentiscus L var chia, which grows only on Chios Island in Greece. CGM is a kind of resin extracted from the stem and leaves, has been used for many centuries in many Mediterranean countries as a dietary supplement and folk medicine for stomach and duodenal ulcers. CGM is known to induce cell cycle arrest and apoptosis in some cancer cells.
This study was undertaken to investigate the alteration of the cell cycle and induction of apoptosis following CGM treatment of HL-60 cells. The viability of the HL-60 cells was assessed using the MTT assay. Hoechst staining and DNA electrophoresis were employed to detect HL-60 cells under- going apoptosis. Western blotting, immunocytochemistry, confocal microscopy, FACScan flow cytometry, MMP activity and proteasome activity analyses were also employed. CGM treatment of HL-60 cells was found to result in a dose- and time-dependent decrease in cell viability and apoptotic cell death. Tested HL-60 cells showed a variety of apoptotic manifestations and induced the downregulation of G1 cell cycle-related proteins. Taken collectively, our present findings demonstrate that CGM strongly induces G1 cell cycle arrest via the modulation of cell cycle-related proteins, and also apoptosis via proteasome, mitochondrial and caspase cascades in HL-60 cells. Hence, we provide evidence that a natural product, CGM could be considered as a novel therapeutic for human leukemia.
Key words : Chios gum mastic (CGM), apoptosis, cell cycle arrest, HL-60 cells.
Introduction
Chios gum mastic (CGM) is produced from Pistiacia lentiscus L var chia, which grows only on Chios Island in Greece. CGM, a kind of resin extracted from the stem and leaves, has been used for many centuries as a dietary supplement and folk medicine for stomach and duodenal ulcers in many Mediterranean countries. He et al. (2007) have demonstrated that CGM treatment inhibited the proliferation of androgen-independent prostate cancer apparently through modulation of the NF-B target gene. In addition, CGM has also been shown to contain compounds that can induce in vitro apoptosis of human colon cancer cells through caspase- dependent pathways (Balan et al., 2007). Our previous report also showed that CGM strongly inhibits cell proliferation by modulating the expression of G1 cell-related proteins and induces apoptosis via the proteasome, mitochondria and caspases cascades in human tongue sqaumous cell carcinoma cells (Lee et al., 2009).
Apoptosis is an essential physiological process required for embryonic development, regulation of immune responses and maintenance of tissue homeostasis. However, apoptosis is also implicated in a wide range of pathological conditions, including immunological diseases, allergy and cancer (Ohta and Yamashita, 1999; Carson and Ribeiro, 1993). The induction of apoptosis leads to specific morphological and biochemical changes, including cell blebbing, exposure of cell surface phosphatidylserine, cell size reduction including cell shrinkage, chromatin condensation and internucleosomal cleavage of genomic DNA (Wyllie et al., 1980; Williams, 1991).
In many studies, HL-60 cell line derived from human promyelocytic leukemia have been widely investigated that the therapeutic efficacy effect of several chemical agents depended on the induction of the cell cycle arrest and apoptosis (Burbano et al., 2009).
*Corresponding author: Bong-Soo Park, Department of Oral Anatomy, School of Dentistry, Pusan National University, Yangsan, 626-870, Korea
Tel.: +82-51-510-8242, Fax.: +82-51-510-8241 E-mail : [email protected]
Recently, it has been reported that some natural medicinal products extracted from herbal plants may enhance apoptosis induction and reduce mitosis in a variety of cancer cells (Park et al., 2005; Kang et al., 2005; Lian et al., 2006; Yoon et al., 2006; Kumagai et al., 2007; Jun et al., 2007; Wu and Lou, 2007). A number of studies have been pursued on the targeted induction of apoptosis to control the unlimited cell growth and proliferation. In addition, induction of apoptosis in the activated cancer cells may be an effective strategic approach for cancer therapy. Moreover, some of recent studies reported that several traditional herbal medicine extracted from herbal plants as Sho-saiko-to, Saposhnikovia divaricata and Salvia miltiorrhiza Bunge induced apoptosis and cell cycle arrest in HL-60 cell line (Sung et al., 1999; Makino et al., 2006; Tai and Cheung, 2007).
Although a few studies elicited the apoptosis-inducing efficacy of CGM on cancer cells in vitro, there is no report about the apoptotic effect of CGM in human leukemia cell lines. The present study was conducted in order to examine the effect of cytotoxicity, the molecular mechanism underlying the expression alterations of cell cycle-related proteins and apoptosis induction in HL-60 human promyelocytic leukemia cell line treated with CGM.
Materials and Methods
Reagents
Chios gum mastic resin was obtained from Mastic Korea (Seoul, Korea). The following reagents were obtained commercially: Suc-LLVY-AMC was from Calbiochem (Darmstadt, Germany); 5,5',6,6'- tetrachloro-1,1',3,3'- tetraethylbenzimidazol carbocyanine iodide (JC-1) was from Molecular Probes (Eugene, OR, USA); RPMI 1640 and fetal bovine serum (FBS) were from Gibco (Gaithersburg, MD, USA); cisplatin, Dimethyl sulfoxide (DMSO), Hoechst 33342, RNase A, proteinase K, aprotinin, leupeptin, phenylmethyl- sulfonyl fluoride (PMSF), thiazolyl blue tetrazolium bromide, collagenase and propidium iodide (PI) were from Sigma (St.
Louis, MO, USA); SuperSignal West Pico enhanced che- miluminescence Western blotting detection reagent was from Pierce (Rockford, IL, USA).
Antibodies
Rabbit polyclonal anti-human AIF antibody was from Upstate (NY, USA); mouse monoclonal anti-human p21WAF1/
CIP1, p27KIP1, caspase-6, caspase-9, caspase-3, caspase-7, Bax, Bcl-2, cytochrome c, DFF45 (ICAD), Cdk2, Cdk4, poly (ADP-ribose) polymerase (PARP), Lamin A/C antibodies, and rabbit polyclonal anti-human -actin antibody, and FITC- conjugated goat anti-mouse and anti-rabbit IgGs were from Santa Cruz Biotechnology (Santa Cruz, CA, USA); Mouse monoclonal anti-human cyclin D3 was from Cell Signaling (Danver, MA, USA); rabbit polyclonal anti-human DFF40 (CAD) antibody was from Stressgen (Ann Arbor, MI,
USA); HRP-conjugated sheep anti-mouse and anti-rabbit IgGs were from Amersham GE Healthcare (Little Chalfont, UK).
Cell culture
The human promyelocytic leuckemic cell line HL-60 (ATCC, Rockville, USA) was cultured in Dulbecco's modified Eagle's medium (DMEM) medium with 4 mM L-glutamine, 1.5 g/L sodium bicarbonate, 4.5 g/L glucose and 1.0 mM sodium pyruvate supplemented with 10% fetal bovine serum (FBS). The cell cultures were maintained and propagated in a humidified 37oC incubator with a 5% CO2 in air atmosphere.
Treatment of Chios gum mastic (CGM)
Cells were cultured on culture dishes and/or several type of wells for 24 h. The original medium was removed and that washed with phosphate-buffered saline (PBS). It was changed that the fresh medium on the plates. CGM (100 mg/ml) stock solution was added to the medium to obtain 5, 10, 25, 50µg/
ml concentrations of the drug. CGM was dissolved in DMSO and it was kept frozen at −20oC until use. The concentrations of DMSO, 0.001-0.2% (vol/vol) used in this study, both as a vehicle for CGM and as a control, had no effect on HL-60 cells proliferation in my preliminary studies.
MTT assay
Cells were placed in a 96-well plate and incubated for 24 h.
The cells were treated with various concentrations and time points of CGM. And then cells were treated with 500µg/ml of MTT stock solution. After the cells were incubated at 37oC with 5% CO2 for 4 h. The medium was aspirated and formed formazan crystals were dissolved in the mixture solution of DMSO and absolute ethanol (1 : 1). Cell viability was monitored on a ELISA reader (Tecan, Mnnedorf, Switzerland) at 570 nm.
Hoechst staining
Cells were harvested and cell suspension was centrifuged onto a clean, fat-free glass slide with a cytocentrifuge. Cells were stained in 4µg/ml Hoechst 33342 for 10 min at 37oC in the dark and fixed for 10 min 4% paraformaldehyde.
DNA electrophoresis
Cells (2× 106) were resuspended in 0.5 ml of lysis buffer [10 mM Tris (pH 7.5), 10 mM EDTA (pH 8.0), 10 mM NaCl and 0.5% SDS] into which proteinase K (200µg/ml) was added. After samples were incubated overnight at 55oC, 200µl of ice cold 5 M NaCl was added and the supernatant containing fragmented DNA was collected after centrifuga- tion. The DNA was then precipitated overnight at −20oC in 50% isopropanol and RNase A-treated for 1 h at 37oC. The DNA from 106 cells (15µl) was equally loaded on each lane of 2% agarose gels in Tris-acetic acid/EDTA buffer containing 0.5 g/ml ethidium bromide at 100 mA for 0.5 h.
Proteasome activity
Cells (1× 106) were lysed in proteasome buffer [10 mM Tris-HCl, pH 7.5, 1 mM EDTA, 2 mM ATP, 20% glycerol, and 4 mM dithiothreitol (DTT)], sonicated, and then centrifuged at 13,000 g at 4oC for 10 min. The supernatant (20µg of protein) were incubated with proteasome activity buffer [0.05 M Tris-HCl, pH 8.0, 0.5 mM EDTA, 50µM Suc- LLVY-AMC] for 1 h 37oC. The intensity of fluorescence of each solution was measured by a modular fluorimetric system (Spex Industries Inc, Edison, NJ, USA) at 380 nm excitatory and 460 nm emission wavelengths. All readings were standardized using the fluorescence intensity of an equal volume of free AMC solution (50µM).
Western blot analysis
Cells (2× 106) treated with CGM were washed twice with ice-cold PBS, resuspended in a RIPA buffer [300 mM NaCl, 50 mM Tris-HCl (pH 7.6), 0.5% TritonX-100, 2 mM PMSF, 2µl/ml aprotinin and 2 µl/ml leupeptin] and incubated at 4oC for 1 h. The lysates were centrifuged at 14,000 revolutions per min for 15 min at 4oC. Protein concentrations of cell lysates were determined with Bradford protein assay (Bio- Rad, Richmond, CA, USA) and 25µg proteins were loaded onto 7.5-15% SDS/PAGE. The gels were transferred to PVDF membrane (Millipore Corporation, Bedford, USA) and reacted with each antibody. Immunostaining with antibodies was performed using SuperSignal West Femto maximum sensitivity substrate and detected with Alpha Imager HP (Alpha Innotech, Santa Clara, CA, USA).
Measurement of mitochondrial membrane potential (MMP)
Cells were plated in a standard 6-well plate at a density of 5× 105/ml. CGM treated cells were incubated for various time points. The cells were harvested and then JC-1 was added directly (1µM final concentration) and incubated for 15 min at 37oC. Flow cytometry to measure MMP (∆ψm) was per- formed on a CYTOMICS FC500 flow cytometry system (Beckman Coulter, Brea, CA USA). The fluroescence emission were collected through a 530/30 band pass filter (FL- 1) and through a 585/42 band pass filter (FL-2), both on a log scale. Data were acquired and analyzed using CXP software version 2.2. The analyzer threshold was adjusted on the FSC channel to exclude noise and most of the subcellular debris.
Immunofluorescent staining
Cells were placed on slides by cytocentrifuge and fixed for 10 min in 4% paraformaldehyde. After blockingnonspecific binding with 3% bovine serum albumin, the cells were incubated with a primary antibody at a dilution of 1 : 100 for 1 h. After the incubation, the cells were washed 3 each for 5 min, and then incubated with FITC-conjugated secondary antibody at a dilution of 1 : 100 for 1 h at room temperature. Fluorescent images were observed and analyzed under Zeiss LSM 510 laser-scanning confocal microscope (Gettingen, Germany).
Quantification of DNA hypoploidy and cell cycle phase by flow cytometry
Cells (2× 106)treated with CGM were incubated for various time points. In each time point, the harvested cells were washed with PBS containing 1% bovine serum albumin and centrifuged at 2,000 rpm for 10 min. The cells were fixed ice- cold 95% ethanol with 0.5% Tween 20 to a final concentration of 70% ethanol. Fixed cells were pelleted, and washed in 1%
BSA-PBS solution. Cells were resuspended in 1 ml PBS containing 20µg/ml RNase A, incubated at 4oC for 30 min, washed once with BSA-PBS, and stained in PI solution (10µg/ml). After cells were incubated at 4oC for 5 min in the dark, DNA content were measured on a CYTOMICS FC500 flow cytometry system (Beckman Coulter, Brea, CA, USA) and data was analyzed using the Multicycle software which allowed a simultaneous estimation of cell-cycle parameters and apoptosis.
Statistical analysis
Three independent experiments were performed, and sta- tistical significance between groups was determined using the paired t-test by SPSS for Win 12.0 for summary data. p < 0.01 and p < 0.05 were considered statistically significant.
Results
Effects of CGM on the viability and proliferation of the HL-60 cells
The cytotoxic effect of CGM was performed to measure the viability of HL-60 cells by MTT assay. After CGM treatment on HL-60 cells (0 to 50µg/ml) at 24 h, the cell viability was reduced at the concentrations of 20 g/ml (77.8%) to 50 g/ml (28.0%) of CGM (Fig. 1A). Also, after treatment of 30µg/ml CGM to the cells, the cell viability was shown in a time- dependent manner (12 h, 68.0%; 24 h, 53.7%; 48 h, 27.9%) (Fig. 1B). Hence, the half maximal inhibitory concentration (IC50) of CGM was at the 30µg/ml for 24 h. This concen- tration was utilized for further assessment of apoptosis and alternation of the cell cycle.
Morphological and biochemical changes in CGM treated HL-60 cells
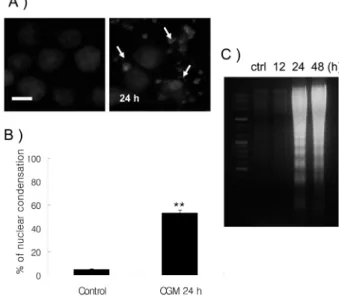
To test whether CGM can change the nuclear morphology of HL-60 cells, Hoechst staining method was performed.
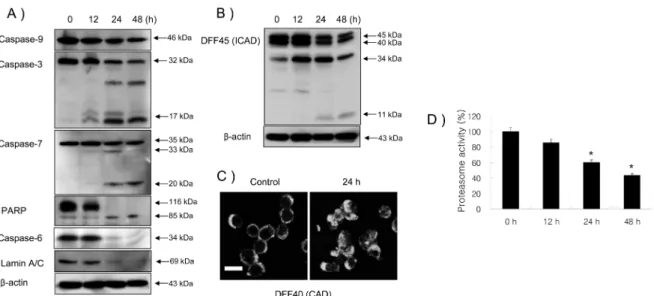
Compared with the typical round nuclei of the control cells, HL-60 cells treated with 30µg/ml CGM for 24 h displayed condensed and fragmented nuclei (Fig. 2A). The conforma- tion value in shape was detected more higher (about 53.0%) than control group (5.0%) (Fig. 2B). DNA fragmentation which is the biochemical hallmark of apoptosis, was demon- strated by DNA electrophoresis. HL60 cells treated with 30µg/ml CGM at 24h and 48 h showed a ladder pattern by DNA electrophoresis (Fig. 2C). As shown by the western blot analysis, CGM treatment at 30µg/ml results in the
induction of active form of caspase-3, caspase-7 and DFF45 (ICAD), and the degradation of caspase-3, caspase-6, caspase- 7, caspase-9, PARP, Lamin A/C and DFF45 (ICAD) in a time- dependent manner (Fig. 3A and 3B). Also, as shown by data in Fig. 3C, CGM led to the translocation of DFF40 (CAD) from cytosol onto the nuclei.
Proteasome activity in HL-60 cells treated with CGM In order to investigate the inhibition effect of proteasome activity at 30µg/ml CGM, proteasome activity assay was employed. In this assay, CGM significantly abolished proteasome activity in a time-dependent manner (Fig. 3D).
Mitochondrial events were closely associated with CGM-induced apoptosis of HL-60 cells
A Western blot assay was performed to examine the role of Bcl-2 family proteins in CGM-induced apoptosis. The expres- sion of Bax was up-regulated in a time-dependent manner, and the expression of Bcl-2 was not down-regulated (Fig. 4A).
The mitochondria were stained with JC-1 dye, and the mitochondria membrane potential (∆ψm) was measured by flow cytometry. HL-60 cells treated with 30µg/ml CGM at various time points showed the loss of mitochondrial
membrane potential (∆ψm) in a time-dependent manner (Fig.
4B). Confocal microscopy was conducted to examine whether AIF and cytochrome c are released in the mitochondria or not.
AIF was translocated to from mitochondria to nuclei and cytochrome c was released from mitochondria into the cytosol in HL-60 cells after treatment of 30µg/ml CGM (Fig. 4C &
4D).
Quantification of DNA hypoploidy in HL-60 cells treated with CGM
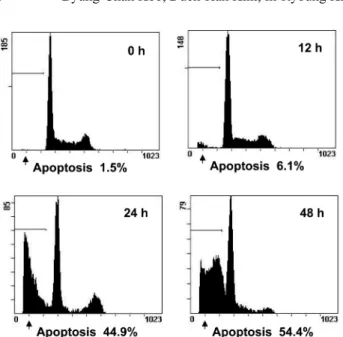
The evaluation of apoptotic percentages was confirmed with flow cytometry analysis. A flow cytometry showed that the treatment of 30µg/m CGM remarkably increased apoptotic cells with DNA hypoploidy in a time-dependent manner compared to the control group (Fig. 5).
The alteration of the cell cycle-related proteins in HL- 60 cells treated with CGM
To investigate the alteration of cell cycle-related proteins, a Western blot assay was conducted. Western blotting data showed that expression level of Cyclin D3, Cdk2 and Cdk4 regulating G1 phase was down-regulated in a time-dependent manner. The expression level of p21WAF1/CIP1 and p27KIP1 was up-regulated in a time-dependent manner (Fig. 6).
Fig. 1. Cytotoxicity of CGM in CGM-treated HL-60 cells as deter- mined by MTT assay (A and B). (A) HL-60 cells were treated with CGM (0~50µg/ml) for 24 h. The viability of HL-60 cells were decreased in a does-dependent manner (20~50µg/ml, p < 0.01).
(B) MTT assay of HL-60 cells treated with 30µg/ml CGM at vari- ous time points. HL-60 cells showed the significant reduction of viability in a time-dependent manner (24 h, p < 0.05; 48 h, p <
0.01). Result is expressed as percentage of the control ± SD of three separate experiments. *, p < 0.05, **, p < 0.01.
Fig. 2. Demonstration of apoptosis in HL-60 cells treated with 30µg/ml CGM. (A) Immunofluorescent micrographs after Hoe- chst staining. Control cells showing round-shape nuclei (left panel). Cells treated with 30µg/ml CGM for 24 h showed the pro- duction of nuclear condensation or fragmentation (right panel).
Note many fragmented nuclei (arrows) (B) The values are the mean ± SD of the mean of apoptotic cells as determined by Hoe- chst staining. The results presented are representatives of three independent experiments *, p < 0.05, **, p < 0.01. (C) DNA frag- mentation was demonstrated by DNA electrophoresis. Cells treated with 30µg/ml CGM for 12 h showed no DNA ladder, but 24 h and 48 h groups showed DNA degradation characteristic of apoptosis with a ladder pattern of DNA fragments.
Fig. 3. Western blot analyses (A and B) of caspase-9, caspase-3, caspase-7, PARP, caspase-6, Lamin A/C and DFF45, confocal microscopy (C), and proteasome activity assay in HL-60 cells treated with 30µg/ml CGM (D). (A) CGM treatment induced caspase-9, caspase-3, caspase-7, PARP, caspase-6, Lamin A/C and DFF45 degradation, and processed caspase-3 17 kDa, caspase-7 33 kDa and 20 kDa, and PARP 85 kDa cleaved products. The levels of β-actin were used as an internal standard for quantifying caspase-9, caspase-3, caspase-7, PARP, caspase-6, Lamin A/C and DFF45 expression. (B) CGM treatment induced the activation of DFF45 (ICAD). DFF45 (ICAD) induced cleaved products (30 kDa and 11 kDa). The levels of β-actin were used as an internal standard for quantifying DFF45 (ICAD) expression. (C) Confo- cal microscopy showing that DFF40 (CAD) translocated from cytosol into nuclei. Scale bar, 10µm. (D) Proteasome activity significantly decreased in a time-dependent manner. Three independent assays were performed. *, p < 0.05, **, p < 0.01.
Fig. 4. Demonstration of mitochondrial events in HL-60 cells after 30 µg/ml CGM treatment. (A) Western blot analysis of Bax and Bcl-2.
Pro-apoptotic factor, Bax significantly up-regulated in a time-dependent manner whereas anti-apoptotic factor, Bcl-2 was not down-regulated.
The levels of β-actin were used as an internal standard for quantifying Bax and Bcl-2 expression. (B) Mitochondrial membrane potential (∆ψm) at various time points. ∆ψm was reduced in HL-60 cells in a time-dependent manner (24 h, p < 0.05; 48 h, p < 0.01). ∆ψm was mea- sured by flow cytometry. Three independent assays were performed. (C) Confocal microscopy showing that cytochrome c was released from mitochondria into the cytosol of HL-60 cells. Scale bar, 10µm. (D) Confocal microscopy showing that AIF was released from mitochondria into the cytosol, and that translocation onto nuclei was evident in HL-60 cells. Scale bar, 10µm.
Discussion
Public attention on natural products as herbal remedies continues to grow. Moreover, the revelation of the pharmacological mechanisms of herbal plant compounds is contributing to their acceptance by healthcare professionals and the public. A number of studies has elucidated that individual herbal medicines extracted from herbal plants
have a number of pharmacological activities, e.g. anti-allergic, anti-pyretic, analgesic, anti-inflammatory and anticancer effects (Kim et al., 1999; Kim et al., 1998; Na et al., 2002; Park et al., 2001; Kim et al., 2003). Furthermore, recent studies have focused on the inhibition of melanogenesis by herbal medicines (Im et al., 2003; Dattner, 2004; Lee et al., 2006).
CGM obtained from the stem and leaves of Pistacia lenticulus trees induces cell cycle arrest and apoptosis in prostate and colon cancer cells (Balan et al., 2007; He et al., 2007). In spite of numerous in vitro and in vivo studies, the mode of action of most herbal medicines remains elusive. Thus, the present work investigating the effects of CGM on cell viability in HL-60 human promyelocytic leukemia cells showed that CGM produced a dose- and time-dependent reduction on viability of HL-60 cells in MTT assay. These data indicate that CGM exerts a cytotoxic effect on HL-60 cells.
Apoptosis and necrosis are conceptually distinct forms of cell death and can be distinguished by their specific morphological changes. During apoptosis, apoptotic cells undergo specific morphological changes such as the cell blebbing, reduction of cell size, cell shrinkage, chromatin condensation and DNA fragmentation (Wyllie et al., 1980; Williams, 1991). In this study, HL-60 cell treated with CGM induced apoptotic hallmarks such as internucleosomal DNA fragmentation and the formation of apoptotic bodies. These results indicate that CGM inhibited the proliferation of HL-60 cells via activation of the apoptosis.
Apoptotic stimuli may induce apoptosis by inhibiting the proteasome activity of the target cells (Meng et al., 1999).
However, other studies have reported that a proteasome inhibitor itself can induce apoptosis in certain cells (Drexler et al., 2000). Generally, the proteasome-mediated apoptosis are located upstream of mitochondrial changes and caspase activation, and can involve different systems, including various cyclins, p53, NF-kB and Bcl-2 family (Orlowski, 1999; Grimm et al., 1996; Li and Dou, 2000). Thus, the possibility existed that CGM may have affected proteasome activity in HL-60 cells and induced the mitochondrial pathway of apoptosis. Our result from proteasome activity assays showed a significant reduction following CGM treatment in a time-dependent manner, suggesting CGM-induced apoptosis via proteasome inhibition.
Mitochondria are known to be central death regulators in response to several apoptotic stimuli (Green and Reed, 1998).
The induction of increased mitochondrial permeability plays a definite role in the regulation of apoptosis (Kroemer et al., 1997; Green and Reed 1988; Susin et al., 1999 Orrenius, 2004). The mitochondrial pathway can also be triggered by various intracellular and extracellular stress signals, which result in activation of pro-apoptotic proteins, including Bax and Bak, or inactivation of anti-apoptotic Bcl-2 family members, such as Bcl-2 and Bcl-xL (Orrenius, 2004). As a result of activation/inactivation of Bcl-2 family proteins, changes in mitochondrial membrane lead to the dissipation of inner membrane potential and permeabilization of the outer Fig. 6. Western blot analysis of cell cycle-related proteins in HL-60
cells treated with 30µg/ml CGM. Cdk2, Cdk4, and Cyclin D3 down-regulated in a time dependent manner. Cdk inhibitors, p21WAF1/CIP1 and p27KIP1 up-regulated in a time-dependent manner.
The levels of β-actin were used as an internal standard for quantify- ing p21WAF1/CIP1, p27KIP1, Cdk2, Cdk4 and Cyclin D3 expression.
Fig. 5. The kinetic analysis of the effect of 30 µg/ml CGM treat- ment on HL-60 cell cycle progression and induction of apoptosis using flow cytometry. Representative DNA histograms are shown.
CGM treatment significantly showed the increase of apoptotic cells with DNA hypoploidy in a time-dependent manner (24 h~48 h, p < 0.01). Data shown are representatives of three independent experiments.
mitochondrial membrane. This in turn induces the release of various proapoptotic proteins, such as cytochrome c, Smac/
Diablo, endonuclease G and AIF (Barczyk et al., 2005;
Brouckaert et al., 2005; Hengartner, 2000). The present study showed the up-regulation of Bax in HL-60 cells treated with CGM. It has been reported that the pro-apoptotic Bcl-2 family in isolated mitochondria induces cytochrome c release, the loss of mitochondrial membrane potential and results in AIF release (Hunter et al., 1996; Narita et al., 1998). Cytochrome c release and disruption of mitochondrial membrane potential (MMP) are known to contribute to apoptosis triggered by proteasome inhibition (Wagenknecht et al., 2000; Marshanskya et al., 2001). Generally, cytochrome c is released into the cytosol during apoptosis, where it binds to Apaf-1. This cytochrome c/Apaf-1 complex (the apoptosome) promotes the autoactivation of procaspase-9 to caspase-9. Caspase-9 then acts on procaspase-3 to initiate a caspase activation cascade (Li et al., 1997; Zou et al., 1999). Released AIF through proapoptotic Bcl-2 family activation induces its translocation to the nucleus, resulting in chromatin condensation and large-scale DNA fragmentation (Douglas et al., 2000). In the present study, CGM treatment also induced translocation of AIF into nuclei, cytochrome c release from mitochondria into the cytosol, significant loss of MMP, and degradation of caspase-9. These results clearly elucidated that the CGM-induced apoptosis in HL-60 cells was involved with mitochondrial events as mentioned above.
Commonly, the final event of apoptosis is nuclear condensation, which is controlled by the known caspase substrates, DFF and PARP (Thornberry and Lazebnik, 1998).
Caspases play an essential role during apoptotic cell death (Acehan et al., 2002). Once activated, the effector caspases (caspase-3, caspase-6 or caspase-7) are responsible for the proteolytic cleavage of a broad spectrum of cellular targets, ultimately leading to cell death. The known cellular substrates include structural components such as actin and nuclear lamin, inhibitors of deoxyribonuclease such as DFF45 (ICAD) and DNA repair proteins such as PARP (Gross et al., 1999; Porter, 1999). In apoptotic cells, activation of DFF40 (CAD), also a substrate of caspase-3, occurs with the cleavage of DFF45 (ICAD). Once DFF40 (CAD) is activated and released from the complex of DFF45 (ICAD) and DFF40 (CAD), it can translocate to the nucleus where it degrades chromosomal DNA and produces DNA fragmentation (Cheng et al., 2007).
In this study, cleavages of caspase-3, caspase-7, DFF45 (ICAD) and PARP, and degradation of caspase-9, -3, -7, -6, DFF45 (ICAD), Lamin A/C and PARP were shown in CGM- treated HL-60 cells. Furthermore, confocal microscopy showed that CGM led to the translocation of DFF40 (CAD) from cytosol into nuclei in HL-60 cells. Therefore, these data demonstrate that CGM-induced apoptosis in HL-60 cells is associated with caspase-9, caspase-3, caspase-7, caspase-6 activation, and that activated caspase-3, caspase-7 and caspase-6 leads to the cleavage and degradation of PARP and DFF45 (ICAD), and the degradation of Lamin A/C, and the
translocation of DFF40 (CAD) from cytosol into nucleus.
Various studies on the molecular analyses of cancers have revealed that cell cycle regulators are frequently mutated in most common malignancies. Therefore, the control of cell cycle progression in tumor cells is considered to be a potentially effective strategy for the inhibition of tumor growth. In the case of Cdks, Cyclins and Cdk inhibitors, these play critical roles in the regulation of cell cycle progression (Pavletich, 1999). Cdk inhibitors inhibit the active Cdk- Cyclin complex (el-Deiry et al., 1994). p21WAF1/CIP1 and p27KIP1 have been demonstrated to play an important role in regulating progression from the G1 phase, by binding to and preventing premature activation of Cdk4/Cyclin D and Cdk2/Cyclin E complexes (Polyak et al., 1994; Coats et al., 1996). The present study demonstrated that CGM inhibited HL-60 cells growth by down-regulation of G1 cell cycle- related proteins and up-regulation of CDK inhibitors. Thus it can be postulated that the mechanism of cell cycle arrest may be associated with up-regulation p21WAF1/CIP1 and p27KIP1, and down-regulation of cyclin D3, Cdk2 and Cdk4.
Therefore, these findings suggest that p21WAF1/CIP1 and p27KIP1 may play a key role as a suppressor in G1 phase arrest on CGM-induced HL-60 cells.
In conclusion, this study demonstrates that CGM strongly inhibits cell proliferation via the alteration of the cell cycle- related proteins including Cyclins, Cdks and Cdk inhibitors, and induces apoptosis via proteasome, mitochondrial and caspase cascades in HL-60 cells. Therefore, our data provide the possibility that a natural product, CGM could be considered as a novel therapeutic strategy for human leukemia.
Acknowledgement
This work was supported by for two years Pusan National University research grant.
References
Acehan D, Jiang X, Morgan DG, Heuser JE, Wang X, Akey CW.
Three-dimensional structure of the apoptosome: Implications for assembly, procaspase-9 binding, and activation. Mol Cell. 2002;9:423-32.
Balan KV, Prince J, Han Z, Dimas K, Cladaras M, Wyche JH, Sitaras NM, Pantazis P. Antiproliferative activity and induction of apoptosis in human colon cancer cells treated in vitro with constituents of a product derived from Pistacia lentiscus L. var. chia. Phytomedicine. 2007;14:263-72.
Barczyk K, Kreuter M, Pryjma J, Booy EP, Maddika S, Ghavami S, Berdel WE, Roth J, Los M. Serum cytochrome c indicates in vivo apoptosis and can serve as a prognostic marker during cancer therapy. Int J Cancer. 2005;21:167-73.
Brouckaert, G, Kalai, M, Saelens, X, Vandenabeele, P.
Apoptotic Pathways and Their Regulation. In: Los, M., Gibson, S.B. (Eds.), Apoptotic Pathways as Target for Novel
Therapies in Cancer and Other Diseases. Springer Academic Press, New York. 2005.
Burbano RR, Lima PD, Bahia MO, Khayat AS, Silva TC, Bezerra FS, Andrade Neto M, de Moraes MO, Montenegro RC, Costa-Lotufo LV, Pessoa C. Cell cycle arrest induced by pisosterol in HL-60 cells with gene amplification. Cell Biol Toxicol. 2009;25:245-51.
Carson DA, Ribeiro JM. Apoptosis and disease. Lancet.
1993;341:1251-4.
Cheng AC, Jian CB, Huang YT, Lai CS, Hsu PC, Pan MH.
Induction of apoptosis by Uncaria tomentosa through reactive oxygen species production, cytochrome c release, and caspases activation in human leukemia cells. Food Chem Toxicol. 2007;45:2206-18.
Coats S, Flanagan WM, Nourse J, Roberts JM. Requirement of p27Kip1 for restriction point control of the fibroblast cell cycle. Science. 1996;272:877-80.
Douglas E, Susin SA, Zamzami N, Ferri KF, Irinopoulou T, Larochette N, Prevost MC, Leber B, Andrews D, Penninger J, Kroemer G. Mitochondrio-nuclear translocation of AIF in apoptosis and necrosis. FASEB J. 2000;14:729-39.
Dattner AM. Herbal and complementary medicine in dermatology. Dermatol Clin. 2004;22:325-32.
Drexler HC, Risau W, Konerding MA. Inhibition of proteasome function induces programmed cell death in proliferating endothelial cells. FASEB J. 2000;14:65-77.
el-Deiry WS, Harper JW, O'Connor PM, Velculescu VE, Canman CE, Jackman J, Pietenpol JA, Burrell M, Hill DE, Wang Y, et al. WAF1/CIP1 is induced in p53-mediated G1 arrest and apoptosis. Cancer Res. 1994;54:1169-74.
Green DR, Reed JC. Mitochondria and apoptosis. Science.
1998;281: 1309-12.
Grimm LM, Goldberg AL, Poirier GG, Schwartz LM, Osborne BA. Proteasome play an essential role in thymocyte apoptosis.
EMBO J. 1996;15:3845-52.
Gross A, McDonnell JM, Korsmeyer SJ. BCL-2 family members and the mitochondria in apoptosis. Genes Dev.
1999;13:1899-911.
He M, Li A, Xu CS, Wang SL, Zhang MJ, Gu H, Yang YQ, Tao HH. Mechanisms of antiprostate cancer by gum mastic: NF- kappaB signal as target. Acta Pharmacol Sin. 2007;28:446-52.
Hengartner, MO. The biochemistry of apoptosis. Nature. 2000;
407:770-6.
Hunter JJ, Parslow TG. A peptide sequence from Bax that converts Bcl-2 into an activator of apoptosis. J Biol Chem.
1996;271:8521-24.
Im SJ, Kim KN, Yun YG, Lee JC, Mun YJ, Kim JH, Woo WH.
Effect of Radix Ginseng and Radix Trichosanthis on the melanogenesis. Biol Pharm Bull. 2003;26:849-53.
Jun DY, Kim JS, Park HS, Han CR, Fang Z, Woo MH, Rhee IK, Kim YH. Apoptogenic activity of auraptene of Zanthoxylum schinifolium toward human acute leukemia Jurkat T cells is associated with ER stress-mediated caspase-8 activation that stimulates mitochondria-dependent or -independent caspase cascade. Carcinogenesis. 2007;28:1303-13.
Kang JX, Liu J, Wnag J, He C, Li FP. The extract of huanglian, a medicinal herb, induces cell growth arrest and apoptosis by
upregulation of interferon-beta and TNF-alpha in human breast cancer cells. Carcinogenesis. 2005;26:1934-9.
Kim HM, Lee EH, Hong SH, Song HJ, Shin MK, Kim SH, Shin TY. Effect of Syzygium aromaticum extract on immediate hypersensitivity in rats. J Ethnopharmacol. 1998;60:125-31.
Kim HM, Yi JM, Lim KS. Magnoliae flos inhibits mast cell- dependent immediate-type allergic reactions. Pharmacol Res. 1999;39:107-11.
Kim JH, Bae HR, Park BS, Lee JM, Ahn HB, Rho JH, Yoo KW, Park WC, Rho SH, Yoon HS, Yoo YH. Early mitochondrial hyperpolarization and intracellular alkalinization in lactacystin- induced apoptosis of retinal pigment epithelial cells. J Pharmacol Exp Ther. 2003;305:474-81.
Kroemer G, Zamzami N and Susin SA. Mitochondrial control of apoptosis. Immunol Today. 1997;18:44-51.
Kumagai T, Müller CI, Desmond JC, Imai Y, Heber D, Koeffler HP. Scutellaria baicalensis, a herbal medicine: anti- proliferative and apoptotic activity against acute lymphocytic leukemia, lymphoma and myeloma cell lines. Leuk Res.
2007;31:523-30.
Lee KJ, Kim JY, Choi JH, Kim HG, Chung YC, Roh SH, Jeong HG. Inhibition of tumor invasion and metastasis by aqueous extract of the radix of Platycodon grandiflorum. Food Chem Toxicol. 2006;44:1890-6.
Lee SE, Hur YJ, Kim IR, Kwak HH, Kim GC, Shin SH, Kim CH, Park BS. Mechanism underlying Chios gum mastic- induced apoptosis on SCC25 human tongue squamous cell carcinoma cell line. Int J Oral Biol. 2009;34:61-77.
Lian Z, Niwa k, Onogi K, Mori H, Harrigan RC, Tamaya T.
Anti-tumor effects of herbal medicines on endometrial carcinomas via estrogen receptor-alpha-related mechanism.
Oncol Rep 2006;15:1133-6.
Li B, Dou QP. Bax degradation by the ubiquitin/proteasome- dependent pathway: Involvement in tumor survival and progression. Pro Natl Acad Sci USA. 2000;97:3850-5.
Li P, Nijhawan D, Budihardjo I, Srinivascula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479-89.
Makino T, Tsubouchi R, Murakami K, Haneda M, Yoshino M.
Generation of reactive oxygen species and induction of apoptosis of HL-60 cells by ingredients of traditional herbal medicine, Sho-saiko-to. Basic Clin Pharmacol Toxicol.
2006;98:401-5.
Marshansky V, Wang X, Bertrand R, Luo H, Duguid W, Chinnadurai G, Kanaan N, Vu MD, Wu J. Proteasomes modulate balance among proapoptotic and antiapoptotic Bcl-2 family members and compromise functioning of the electron transport chain in leukemic cells. J Immunol. 2001;166:3130- 42.
Meng L, Kwok BH, Sin N, Crews CM. Eponemycin exerts its antitumor effect through the inhibition of proteasome function. Cancer Res. 1999;59:2798-801.
Na HJ, Jeong HJ, Bae H, Kim YB, Park ST, Yun YG, Kim HM.
Tongkyutang inhibits mast cell-dependent allergic reactions and inflammatory cytokines secretion. Clin Chim Acta.
2002;319:35-41.
Narita M, Shimizu S, Ito T, Chittenden T, Lutz RJ, Matsuda H,
Tsujimoto Y. Bax interacts with the permeability transition pore to induce permeability transition and cytochrome c release in isolated mitochondria. Proc Natl Acad Sci USA.
1998;95:14681-6.
Ohta K, Yamashita N. Apoptosis of eosinophils and lymphocytes in allergic inflammation. J Allergy Clin Immunol. 1999;104:
14-21.
Orlowski RZ. The role of the ubiquitin-proteasome pathway in apoptosis. Cell Death Differ. 1999;6:303-13.
Orrenius S. Mitochondrial regulation of apoptotic cell death.
Toxicol Lett. 2004;149:19-23.
Park BS, Song YS, Yee SB, Lee BJ, Seo SY, Park YC, Kim JY, Kim HM, Yoo YH. Phospho-ser 15-p53 translocate into mitochondria and interacts with Bcl-2 and Bcl-xL in eugenol- induced apoptosis. Apoptosis. 2005;10:193-200.
Park HI, Jeong MH, Lim YJ, Park BS, Kim GC, Lee YM, Kim HM, Yoo KS, Yoo YH. Szygium aromaticum (L.) Merr. Et Perry (Myrtaceae) flower bud induces apoptosis of p815 mastocytoma cell line. Life Sci. 2001;69:553-66.
Pavletich NP. Mechanisms of cyclin-dependent kinase regulation:
structures of Cdks, their cyclin activators, and Cip and INK4 inhibitors. J Mol Biol. 1999;287:821-8.
Polyak K, Lee MH, Erdjument-Bromage H, Koff A, Roberts JM, Tempst P, Massagué J. Cloning of p27Kip1, a cyclin- dependent kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell. 1994;78:59-66.
Porter AG. Protein translocation in apoptosis. Trends Cell Biol.
1999;9:394-401.
Sung HJ, Choi SM, Yoon Y, An KS. Tanshinone IIA, an ingredient of Salvia miltiorrhiza BUNGE, induces apoptosis
in human leukemia cell lines through the activation of caspase-3. Exp Mol Med. 1999;31:174-8.
Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM, Kroemer G. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:
441-6.
Tai J and Cheung S. Anti-proliferative and antioxidant activities of Saposhnikovia divaricata. Oncol Rep. 2007;18:227-34.
Thornberry NA and Lazebnik Y. Caspase: Enemies within.
Science. 1998;28: 1312-6.
Wagenknecht B, Hermission M, Groscurth P, Liston P, Krammer PH, Weller M. Proteasome inhibitor-induced apoptosis of glioma cells involves the processing of multiple caspases and cytochrome c release. J Neurochem. 2000;75:2288-97.
Williams GT. Programmed cell death: apoptosis and oncogenesis.
Cell 1991;65: 1097-8.
Wu YD, Lou YJ. A steroid fraction of chloroform extract from bee pollen of Brassica campestris induces apoptosis in human prostate cancer PC-3 cells. Phytother Res. 2007;21:1087-91.
Wyllie AH, Kerr JF, Currie AR. Cell death: the significance of apoptosis. Int Rev Cytol 1980;68:251-306.
Yoon JS, Seo JC, Han SW. Pinelliae Rhizoma herbal- acupuncture solution induced apoptosis in human cervical cancer cells, SNU-17. Am J Chin Med. 2006;34:401-8.
Zou H, Li Y, Liu X, Wang, X. An APAF-1, cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J. Biol Chem. 1999;274:11549-56.