Received: June 1, 2016 / Accepted: June 8, 2016 Address for correspondence: Hyung Jun Park MD
유전성근육질환의 임상적 접근
이화여자대학교 의과대학 목동병원 신경과
박 형 준
Clinical Approach to Inherited Muscular Disorders
Hyung Jun Park, MD
Department of Neurology, Mokdong Hospital, Ewha Womans University School of Medicine, Seoul, Korea
KEYWORDS
Inherited muscular disorder, Muscular dystrophy, Phenotype, Diagnosis
Inherited muscular disorders (IMDs) are clinically and genetically heterogeneous groups of genetic disorders characterized by skeletal muscle weakness. These genetic and phenotypic complexities of IMDs make it difficult to identify the causative genes by a classical diagnostic approach, using clinical, pathological and genetic analysis in sequential steps. The recent advance in next gen- eration sequencing makes it possible to sequence many genes cost-effectively and rapidly and has improved the molecular diagnosis of IMDs. However, comprehensive analysis of clinical phenotype is still important and required to identify pathogenic variants among large genomic data including non-pathogenic variants and sequencing errors. In this article, we provide a comprehensive review that integrates clinical manifestations and diagnostic strategy.
서 론
유전성근육질환은 골격근의 약화를 특징으로 하는 임상 표현형과 유전적 원인이 다양한 유전질환이다. 유전성근 육질환의 원인으로 현재까지 130개 이상의 유전자가 밝혀 졌다.1 유전성근육질환은 임상적 특징과 병리학적 소견에 따라서 근육디스트로피(muscular dystrophy), 선천성근육 디스트로피(congenital muscular dystrophy), 원위부근육병 (distal myopathy), 선천성근육병(congenital myopathy), 대 사근육병(metabolic myopathy), 통로병증(channelopathy) 등으로 구분된다. 그러나 이러한 유전성근육질환의 아형 들 간에도 임상양상 및 병리소견의 유사성이 존재하고, 원 인유전자가 같은 경우도 많아서 아형을 구분하는 것도 쉬
운 일이 아니다. 이러한 유전학적 그리고 임상적 다양성은 표현형분석/병리소견분석/단백발현확인/유전자검사의 순 으로 진행하는 고전적인 유전성근육질환의 진단방법의 가 장 큰 장애물이었다. 최근 차세대염기서열이 도입으로 많 은 유전자들을 빠르고 효과적으로 분석할 수 있게 되어서 유전성근육질환의 진단율은 획기적으로 높아졌다.2,3 그러 나 유전성근육질환에서의 표현형 분석은 여전히 가장 중 요한 작업 중 하나로 차세대염기서열분석으로 찾은 많은 비병원성 변이들 중 원인유전자를 결정하는 데 꼭 필요하 다.2-4 본 논문에서는 유전성근육질환에서 유전학적 원인 에 따른 임상표현형과 병리학적 특징을 정리하고, 임상적 인 접근방법에 대해서 살펴보기로 하겠다.
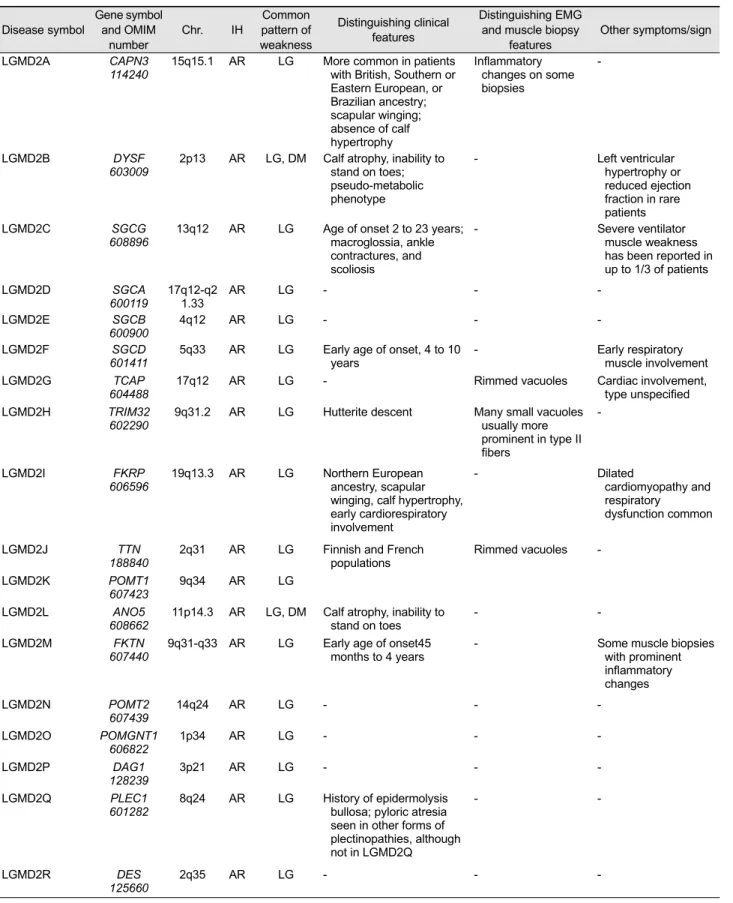
number weakness features features Muscular dystrophy
DMD/BMD DMD
300377
Xp21.2 XR LG Calf hypertrophy Mosaic appearance of dystrophin on immunohistochemist ry in affected woman
Dilated
cardiomyopathy
EDMD-X1 END
300384
Xq28 XR HP Contractures invariably present early in the disease course
- Cardiac conduction
abnormalities
EDMD-X2 FHL1
300163
Xq27.2 XR LG, HP or SP, DM
Foot drop; extremity contractures and neck contractures common
Myofibrillar myopathy or reducing bodies
Severe respiratory failure in many patients EDMD, AD;
EDMD, AR
LMNA 150330
1q21.2 AD /AR
LG, HP, DM
Early humeroperoneal weakness, limb contractures
Cardiomyopathy and conduction system disease
FSHD type I DUX4
606009
4q35 AD FSH, SP Asymmetric
facioscapulohumeral muscle weakness
- -
FSHD type II SMCHD1 614982
18p11.32 AD FSH, SP Asymmetric
facioscapulohumeral muscle weakness
- -
Myotonic dystrophy
DMPK 605377
19q13 AD DM Clinical myotonia, distal muscle weakness
Myotonic discharges A long, thin face with hollow temples and drooping eyelids;
Diabetes
Limb-girdle muscular dystrophy
LGMD1A MYOT
604103
5q31 AD LG, DM Onset>40 years, foot drop, asymmetric muscle weakness and atrophy
Myofibrillar myopathy, myotonic or pseudomyotonic discharges
Cardiomyopathy, respiratory muscle weakness
LGMD1B LMNA 1q21.2 AD LG, HP,
DM
Early humeroperoneal weakness, limb contractures
Cardiomyopathy and conduction system disease
LGMD1C CAV3
601253
3p25 AD LG Rippling muscles, percussion-induced rapid contractions, prominent muscle cramp and calf hypertrophy
- -
LGMD1D DNAJB6
611332
7q36.2 AD LG, DM Onset>40 years, foot drop Myofibrillar myopathy, rimmed vacuoles, myotonic or pseudomyotonic discharges
-
LGMD1E DES
125660
2q35 AD LG, HP, DM
Onset<40 years, foot drop Myofibrillar myopathy, rimmed vacuoles, myotonic or pseudomyotonic discharges
Cardiomyopathy, respiratory muscle weakness
LGMD1F TNPO3
610032
7q32.1-q 32.2
AD LG -
LGMD1G HNRPDL
607137
4q21 AD LG
Table 1. Continue Disease symbol
Gene symbol and OMIM
number
Chr. IH
Common pattern of weakness
Distinguishing clinical features
Distinguishing EMG and muscle biopsy
features
Other symptoms/sign
LGMD2A CAPN3
114240 15q15.1 AR LG More common in patients with British, Southern or Eastern European, or Brazilian ancestry;
scapular winging;
absence of calf hypertrophy
Inflammatory changes on some biopsies
-
LGMD2B DYSF
603009
2p13 AR LG, DM Calf atrophy, inability to stand on toes;
pseudo-metabolic phenotype
- Left ventricular
hypertrophy or reduced ejection fraction in rare patients
LGMD2C SGCG
608896
13q12 AR LG Age of onset 2 to 23 years;
macroglossia, ankle contractures, and scoliosis
- Severe ventilator
muscle weakness has been reported in up to 1/3 of patients
LGMD2D SGCA
600119 17q12-q2
1.33 AR LG - - -
LGMD2E SGCB
600900
4q12 AR LG - - -
LGMD2F SGCD
601411
5q33 AR LG Early age of onset, 4 to 10 years
- Early respiratory
muscle involvement
LGMD2G TCAP
604488
17q12 AR LG - Rimmed vacuoles Cardiac involvement,
type unspecified
LGMD2H TRIM32
602290 9q31.2 AR LG Hutterite descent Many small vacuoles usually more prominent in type II fibers
-
LGMD2I FKRP
606596 19q13.3 AR LG Northern European ancestry, scapular winging, calf hypertrophy, early cardiorespiratory involvement
- Dilated
cardiomyopathy and respiratory
dysfunction common
LGMD2J TTN
188840
2q31 AR LG Finnish and French populations
Rimmed vacuoles -
LGMD2K POMT1
607423
9q34 AR LG
LGMD2L ANO5
608662 11p14.3 AR LG, DM Calf atrophy, inability to
stand on toes - -
LGMD2M FKTN
607440 9q31-q33 AR LG Early age of onset45
months to 4 years - Some muscle biopsies
with prominent inflammatory changes
LGMD2N POMT2
607439
14q24 AR LG - - -
LGMD2O POMGNT1
606822
1p34 AR LG - - -
LGMD2P DAG1
128239 3p21 AR LG - - -
LGMD2Q PLEC1
601282 8q24 AR LG History of epidermolysis bullosa; pyloric atresia seen in other forms of plectinopathies, although not in LGMD2Q
- -
LGMD2R DES
125660
2q35 AR LG - - -
number weakness features
features
LGMD2S TRAPPC11
614138
4q35.1 AR LG - - -
LGMD2T GMPPB
615320
3p21.31 AR LG - - -
LGMD2U ISPD
614631
7p21.2 AR LG - - -
LGMD2V GAA
606800
17q25 AR LG - Glycogan storage
disease
Respiratory failure
Congenital muscular dystrophy CMD with
merosin deficiency (MDC1A)
LAMA2 156225
6q2 AR Sitting and standing with support as maximal motor ability if complete deficiency,
- Abnormal white matter
signal (T2 MRI);
neuropathy, epilepsy, subclinical
cardiomyopathy Ullrich CMD COL6A1
120220, COL6A2 120240 COL6A3
120250
21q22.3, 21q22.3, 2q37
AR LG, HP Distal joint hyperextensibility, proximal contractures, motor abilities variable, precludes independent ambulation in severe cases, soft palmar skin
- -
Bethlem myopathy
COL6A1 120220, COL6A2 120240 COL6A3
120250
21q22.3, 21q22.3, 2q37
AD LG, HP Joint contracture, variable age of onset
- -
Fukutin related proteinopathy (MDC1C)
FKRP 606596
19q13.3 AR LG Often reminiscent of MDC1A, but severity more variable
- Range from normal to
significant brain structural abnormalities LARGE related
CMD (MDC1D)
LARGE 603590
22q12 AR Congenital muscular
dystrophy with profound mental retardation may eventually blend with the MEB/WWS spectrum
- White matter changes,
hypoplastic brainstem, mild pachygyria
Fukuyama CMD FKTN 607440
9q31-q33 AR Frequent Japanese
population, never walk, mental retardation, epilepsy common
- Lissencephaly type
II/pachygyria, hypoplastic brainstem CMD related to
integrin
ITGA7 600536
12q13 AR Very rare, delayed motor milestones, walking at 2-3years
- No CNS involvement
Rigid spine syndrome
SEPN1 606210
1p36 AR LG Rigid spine, joint
contractures, generalized weakness
Multi-mini cores, cores, Mallory bodies, type I fiber predominance
Restrictive lung disease
Myofibrillar myopathy αB-Crystallin CRYAB
123590
11q22 AD LG, DM Early or late onset, foot drop
Myofibrillar myopathy, myotonic or pseudomyotonic discharges
Cardiomyopathy, respiratory muscle weakness
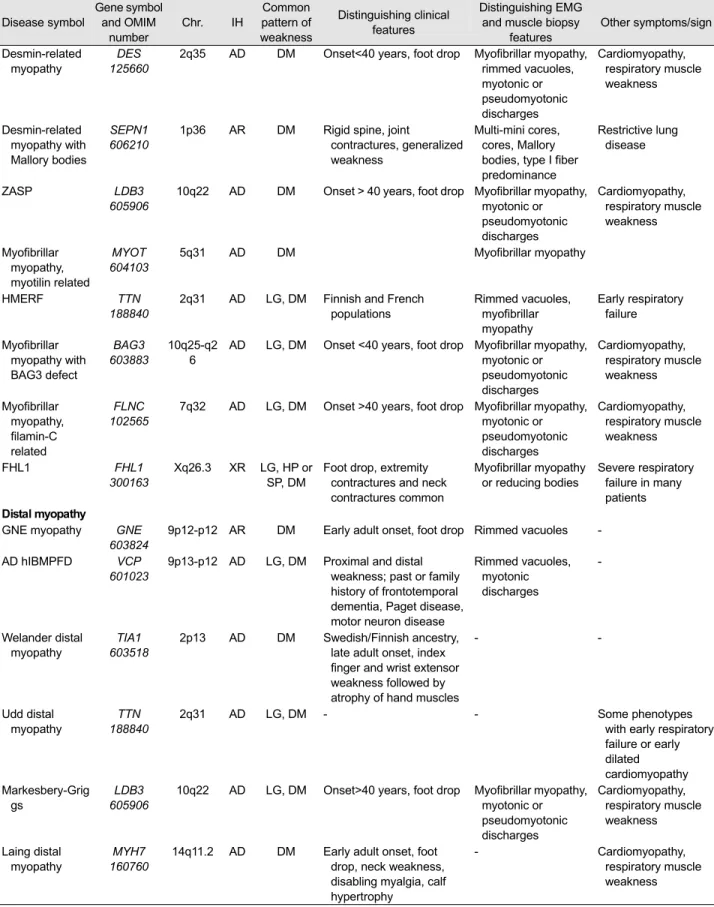
Table 1. Continue Disease symbol
Gene symbol and OMIM
number
Chr. IH
Common pattern of weakness
Distinguishing clinical features
Distinguishing EMG and muscle biopsy
features
Other symptoms/sign
Desmin-related myopathy
DES 125660
2q35 AD DM Onset<40 years, foot drop Myofibrillar myopathy, rimmed vacuoles, myotonic or pseudomyotonic discharges
Cardiomyopathy, respiratory muscle weakness
Desmin-related myopathy with Mallory bodies
SEPN1 606210
1p36 AR DM Rigid spine, joint
contractures, generalized weakness
Multi-mini cores, cores, Mallory bodies, type I fiber predominance
Restrictive lung disease
ZASP LDB3
605906
10q22 AD DM Onset > 40 years, foot drop Myofibrillar myopathy, myotonic or pseudomyotonic discharges
Cardiomyopathy, respiratory muscle weakness
Myofibrillar myopathy, myotilin related
MYOT 604103
5q31 AD DM Myofibrillar myopathy
HMERF TTN
188840
2q31 AD LG, DM Finnish and French populations
Rimmed vacuoles, myofibrillar myopathy
Early respiratory failure
Myofibrillar myopathy with BAG3 defect
BAG3 603883
10q25-q2 6
AD LG, DM Onset <40 years, foot drop Myofibrillar myopathy, myotonic or pseudomyotonic discharges
Cardiomyopathy, respiratory muscle weakness
Myofibrillar myopathy, filamin-C related
FLNC 102565
7q32 AD LG, DM Onset >40 years, foot drop Myofibrillar myopathy, myotonic or pseudomyotonic discharges
Cardiomyopathy, respiratory muscle weakness
FHL1 FHL1
300163
Xq26.3 XR LG, HP or SP, DM
Foot drop, extremity contractures and neck contractures common
Myofibrillar myopathy or reducing bodies
Severe respiratory failure in many patients Distal myopathy
GNE myopathy GNE
603824
9p12-p12 AR DM Early adult onset, foot drop Rimmed vacuoles -
AD hIBMPFD VCP
601023
9p13-p12 AD LG, DM Proximal and distal weakness; past or family history of frontotemporal dementia, Paget disease, motor neuron disease
Rimmed vacuoles, myotonic discharges
-
Welander distal myopathy
TIA1 603518
2p13 AD DM Swedish/Finnish ancestry, late adult onset, index finger and wrist extensor weakness followed by atrophy of hand muscles
- -
Udd distal myopathy
TTN 188840
2q31 AD LG, DM - - Some phenotypes
with early respiratory failure or early dilated cardiomyopathy Markesbery-Grig
gs
LDB3 605906
10q22 AD LG, DM Onset>40 years, foot drop Myofibrillar myopathy, myotonic or pseudomyotonic discharges
Cardiomyopathy, respiratory muscle weakness
Laing distal myopathy
MYH7 160760
14q11.2 AD DM Early adult onset, foot drop, neck weakness, disabling myalgia, calf
- Cardiomyopathy,
respiratory muscle weakness
number weakness features
features Vocal cord and
pharyngeal weakness with distal myopathy
MATR3 164015
5q31 AD DM Mean age at onset 45 years, foot drop and distal upper extremity
weakness, dysphagia, dysphonia
Subsarcolemmal rimmed vacuoles
-
Distal myopathy with Kelch-like homologue 9 mutations
KLHL9 611201
9p22 AD DM Distal weakness - -
Miyoshi myopathy
DYSF 603009
2p12-14 AR DM Calf atrophy, inability to stand on toes
- Left ventricular
hypertrophy or reduced ejection fraction in rare patients Miyoshi
myopathy type III
ANO5 608662
11p14-12 AR DM Calf atrophy, inability to stand on toes
- -
Nebulin myopathy
NEB 161650
2q22 AR DM Juvenile onset, foot drop Nemaline rods -
Dynamin 2 related distal myopathy
DNM2 602378
19p13.2 AD DM Early adult onset, foot drop Central nucleus -
AD, autosomal dominant; AR, autosomal recessive; BMD, Becker muscular dystrophy; Chr, chromosome; DM, distal muscular; DMD, Duchenne muscular dystrophy; EDMD, Emery-Dreifuss muscular dystrophy; FSH, facio-scapulohumeral; FSHD, facio-scapulohumeral muscular dystrophy; hIBMPFD, hereditary inclusion body myopathy with Paget disease and frontotemporal dementia; HMERF, hereditary myopathy with early respiratory failure; HP, humeroperoneal; IH, inheritance; LG, limb-girdle; MEB, muscle eye brain disease; PTRF, polymerase I and transcript factor; SP, scapuloperoneal; WWS, Walker-Warburg syndrome; XR, X-linked recessive.
본 론
근육디스트로피
근육디스트로피는 진행하는 팔다리의 근력약화라는 임상적 특징과 퇴행세포(degenerative cell)와 재생세포(regenerative cell)를 특징적 병리소견을 특징으로 하는 유전성근육질환 이다.5 근육디스트로피는 전통적으로 임상양상과 증상 발 현 나이에 따라서 팔다리이음근육디스트로피(limb-girdle muscular dystrophy, LGMD), 에머리-드레이푸스근육디스 트로피(Emery-Dreifuss muscular dystrophy), 선천성근육디 스트로피(congenital muscular dystrophy)로 분류한다. 이 들은 다시 원인유전자나 유전좌(locus)에 따라 구분되는데 팔다리이음근육디스트로피의 경우 상염색체 우성유전을 1형과 상염색체 열성유전인 2형으로 우선 분류하고, 원인 유전자 또는 유전좌가 밝혀진 순서에 따라서 A, B, C의 알파벳으로 접미사를 붙여서 명명하고 있다.
근육디스트로피에서 소아에서 가장 흔한 질환은 듀센형 근육디스트로피(Duchenne muscular dystrophy, DMD)와 베커근육디스트로피(Becker muscular dystrophy, BMD)이 다. 이 두 질환은 모두 디스트로핀(dystrophin) 유전자의
돌연변이가 원인이고, 유병률은 출생남아 3,500명당 1명 이며, 북잉글랜드의 경우 DMD의 유병률은 출생남아 100,000명당 8.29명, BMD의 유병률은 조금 낮은 100,000 명 소년 중 7.29명으로 알려져 있다.5 반면, 성인에서 가 장 흔한 근육디스트로피는 얼굴어깨위팔근육디스트로피 (facioscapulohumeral muscular dystrophy)와 근긴장디스트 로피(myotonic dystrophy)로 유병률이 각각 100,000명당 12명과 100,000명당 10.6명으로 보고되고 있다.6 팔다리이 음근육디스트로피의 경우 상염색체열성유전의 경우가 상 염색체우성유전의 경우보다 많고, 원인유전자 별로 보면 남유럽에서는 LGMD2A이 북유럽에서는 LGMD2I가 가장 많고, LGMD2B가 두 번째로 많은 것으로 알려져 있다.7-9 선천성근육디스트로피도 원인유전자의 빈도가 지역마다 달라서 일본의 경우는 FKTN 유전자가 주된 원인유전자이 나 대부분의 지역에서는 COL6A1, COL6A2, COL6A3 유 전자의 이상이 원인이 되는 울리히선천성근육디스트로피 (Ulrich congenital muscular dystrophy)가 가장 많은 아형 이다. Table 1은 다양한 근육디스트로피의 원인유전자와 임상양상 중 대표적인 근육디스트로피의 아형과 임상표현 형 및 병리학적 특징을 정리한 것이다.
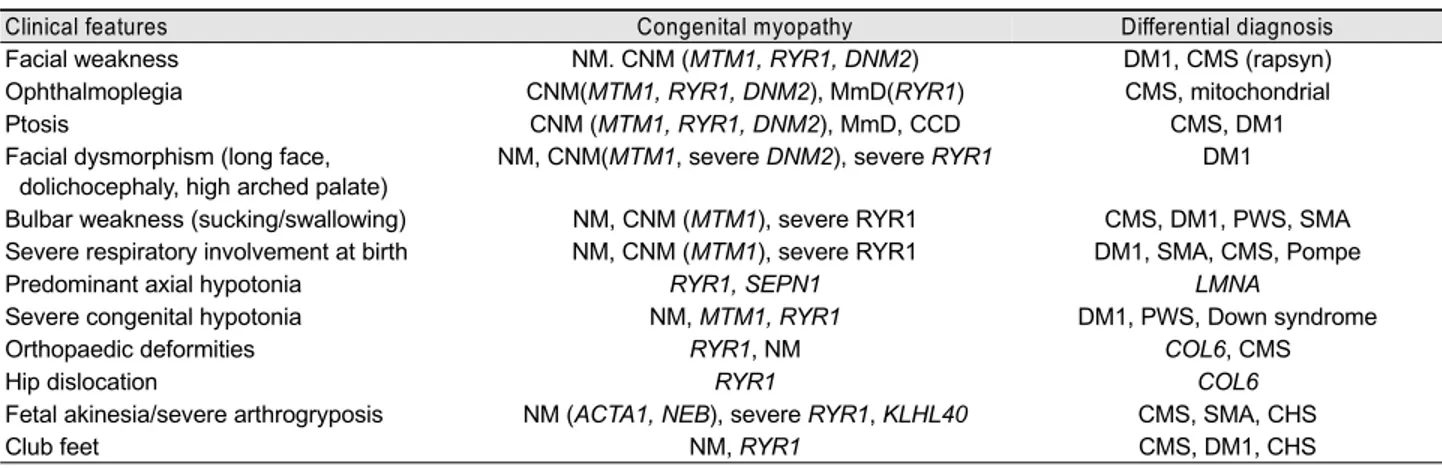
Table 2. Clinical evidences suggestive of specific diagnosis in congenital myopathies: newborn and infant <2 years
Clinical features Congenital myopathy Differential diagnosis
Facial weakness NM. CNM (MTM1, RYR1, DNM2) DM1, CMS (rapsyn)
Ophthalmoplegia CNM(MTM1, RYR1, DNM2), MmD(RYR1) CMS, mitochondrial
Ptosis CNM (MTM1, RYR1, DNM2), MmD, CCD CMS, DM1
Facial dysmorphism (long face, dolichocephaly, high arched palate)
NM, CNM(MTM1, severe DNM2), severe RYR1 DM1
Bulbar weakness (sucking/swallowing) NM, CNM (MTM1), severe RYR1 CMS, DM1, PWS, SMA Severe respiratory involvement at birth NM, CNM (MTM1), severe RYR1 DM1, SMA, CMS, Pompe
Predominant axial hypotonia RYR1, SEPN1 LMNA
Severe congenital hypotonia NM, MTM1, RYR1 DM1, PWS, Down syndrome
Orthopaedic deformities RYR1, NM COL6, CMS
Hip dislocation RYR1 COL6
Fetal akinesia/severe arthrogryposis NM (ACTA1, NEB), severe RYR1, KLHL40 CMS, SMA, CHS
Club feet NM, RYR1 CMS, DM1, CHS
Please note that the table is meant to indicate if a clinical finding is a particular clue to one of the congenital myopathies. Specific clinical findings can occur in the other forms of congenital myopathy at lower frequency.
NM, nemaline myopathy; DM1, myotonic dystrophy type 1; CMS, congenital myasthenic syndrome; CNM, centronuclear myopathy; mito, mitochondrial myopathy; MmD, multi-minicore disease; CCD, central core disease; PWS, Prader Willi syndrome; SMA, spinal muscular atrophy; Pompe, Pompe disease; COL6, collagen VI associated myopathy.
Table 3. Clinical evidences suggestive of specific diagnosis in congenital myopathies: older child
Clinical features Congenital myopathy Differential diagnosis
Scoliosis SEPN1, RYR1, NM COL6, LAMA2
Rigid spine SEPN1, RYR1
Cardiomyopathy TTN, MYH7, rarely ACTA1 Pompe disease
Foot drop/pes cavus NM (NEB, TPM3, TPM2), DNM2, MYH7 Peripheral neuropathy
Malignant hyperthermia CCD, MmD and CNM (RYR1 only)
Respiratory involvement and axial involvement out of proportion to skeletal muscle weakness
SEPN1, NM (NEB, TPM3, ACTA1) LMNA, CMS, Pompe disease
Please note that the table is meant to indicate if a clinical finding is a particular clue to one of the congenital myopathies. Specific clinical find- ings can occur in the other forms of congenital myopathy at lower frequency.
선천성근육병
선천성근육병은 출생 시부터 저긴장증과 근력약화가 있 지만 임상증상의 진행은 없거나 매우 느린 유전성근육질 환이다.10 선천성근육병은 전통적으로 근육조직검사상 구 조적 특징을 기준으로 분류하고 있다. 근육조직검사상의 대표적인 구조적 특징은 네말린근육병(nemaline myo- pathy)에서 보이는 막대, 중심코어근육병(central core dis- ease)과 여러작은코어근육병(multimini core disease)에서 보 이는 코어, 중심핵근육병(centronuclear myopathy)의 중심 핵, 선천성섬유형불균형(congenital fiber type disproportion) 에서 보이는 1형근육섬유의 선택적 위축이 대표적이다.
그러나 최근 분자유전학적 발달로 원인유전자가 밝혀지면 서 이러한 병리소견에 따른 분류법의 제한점이 드러나고 있다. 첫째, 같은 구조적 특징이 있는 근육병 내에서도 원 인유전자가 매우 다양하다. 네말린근육병의 경우 현재까 지 밝혀진 원인유전자만 8개이다. 둘째, 같은 원인유전자 이상을 갖는 근육병에서도 병리소견은 서로 다른 구조적
이상을 보이는 경우가 있다. ACTA1 유전자의 돌연변이로 인한 근육병에서 병리소견 상으로는 네말린근육병, 핵내 막대근육병(intranuclear rod myopathy), 선천성섬유형불균 형, 엑틴축적(actin accumulation), 캡근육병(cap disease), 제브라몸통근육병(zebra body disease)을 갖는 것이 보고
되었다.11-15 셋째, 같은 원인유전자의 돌연변이를 갖는 같
은 가족 내의 환자에서 조차도 병리소견상으로는 서로 다 른 구조적 이상이 관찰할 수 있었다. 이것은 RYR1 유전자 의 돌연변이를 갖는 가족에서 보고된 바 있다.16
선천성근육병의 임상증상은 출생 시부터 심한 근긴장저 하증을 보이는 아기부터 전체적인 근력약화를 갖는 소아 나 장년기에 되어서야 몸통 쪽 근력약화를 보이는 경우까 지 그 정도가 매우 다양하다. 일반적으로는 몸통 쪽 근육 약화와 근위축이 주된 임상소견이지만, 척추주변근육 및 호흡근육 또는 발목올림근육의 약화를 보이는 경우도 있 다. 혈청 크레아틴 키나아제(creatine kinase)는 일반적으로 정상이거나 조금 높은 경우가 대부분이어서 만약 유전성
KBTBD13, KLHL40
Cores RYR1, SEPN1, ACTA1, TTN, MYH7, KBTBD13 Core myopathies
Central nuclei MTM1, DNM2, BIN1, RYR1, (DM1) Centronuclear myopathies
Rods and cores RYR1, NEB, KBTBD13, CFL2 Core-rod myopathies
Caps TPM2, TPM3, ACTA1 Cap disease
Congenital fibre type disproportion ACTA1, TPM3, TPM2, RYR1, SEPN1
Distal myopathy no rods NEB
Distal arthrogryposis TPM2, MYH3, MYH8, TNNT1, TNNT2, TNNT3 DM1, myotonic dystrophy type 1.
근육병 환자에서 혈청 크레아틴 키나아제 수치가 정상 상 한치의 5배 이상 높다면 선천성근육병보다는 근육디스트 로피가 의심되는 소견이다.10 또한 근전도검사에서는 근육 병에 합당한 소견 또는 정상 소견이 주로 관찰된다. 그러 나 때때로 신경병을 시사하는 소견도 보이는데 주로 신생 아기의 심한 근력소실이 있을 경우나 질환이 진행된 후 먼쪽 근육에서 관찰된다.17 Table 2, 3은 각각 신생아기와 소아기로 나누어서 선천성근육병의 임상증상에 따라서 고 려할 수 있는 아형 및 원인유전자를 정리한 것이다. 또한 Table 4는 선천성근육병 환자의 병리소견에 따라 고려해 야 할 원인유전자를 정리한 것이다.
결 론
유전성근육질환은 대표적인 희귀난치성 질환으로 유전 학적 최종진단까지 도달하기가 매우 어렵고, 진단이 되어 도 대부분의 경우 보존적 치료법외에 치료약제가 없는 경 우가 많다. 그러나 게놈프로젝트의 성공과 차세대염기서 열분석의 적용과 같은 분자유전학적 발달은 유전성근육질 환의 유전학적 진단을 보다 경제적이고 효율적으로 개선 시켰고, 치료제 개발에서도 부분적 성공을 거두었으며 현 재도 많은 임상시험들이 이루어지고 있다. 또한 유전학적 원인만 알 수 있다면 착상전진단(preimplantation genetic diagnosis)을 통해서 질환의 유전을 차단하는 것은 지금도 가능하다. 비록 유전성근육질환의 진단에서 임상의사의 역할인 임상표현형분석, 병리소견분석과 같은 임상적 접 근의 중요성이 예전보다는 떨어졌다고 생각할 수 있지만, 막대한 양의 유전정보 중 하나의 원인유전자를 규명하기 위해서 여전히 가장 중요하고 필요한 작업이다. 따라서, 진단과 치료가 괄목할만한 성장을 하고 있는 지금이야 말 로 임상가로서 유전성근육질환의 임상표현형에 주의를 기 울이고 최종 진단에 도달하기 위해서 노력해야 할 시기라 고 생각한다.
REFERENCES
1. Kaplan JC, Hamroun D. The 2016 version of the gene table of monogenic neuromuscular disorders (nuclear genome). Neuromuscul Disord 2015;25:991-1020.
2. Sevy A, Cerino M, Gorokhova S, Dionnet E, Mathieu Y, Verschueren A, et al. Improving molecular diagnosis of distal myopathies by targeted next-generation sequencing. J Neurol Neurosurg Psychiatry 2016;87:235-243.
3. Kuhn M, Gläser D, Joshi PR, Zierz S, Wenninger S, Schoser B, et al. Utility of a next-generation sequencing-based gene panel in- vestigation in German patients with genetically unclassified limb-girdle muscular dystrophy. J Neurol 2016;263:743-750.
4. Ghaoui R, Cooper ST, Lek M, Jones K, Corbett A, Reddel SW, et al. Use of Whole-Exome Sequencing for diagnosis of limb-girdle muscular dystrophy: outcomes and lessons learned. JAMA Neurol 2015;72:1424-1432.
5. Mercuri E, Muntoni F. Muscular dystrophies. Lancet 2013;
381:845-860.
6. Deenen JC, Arnts H, van der Maarel SM, Padberg GW, Verschuuren JJ, Bakker E, et al. Population-based incidence and prevalence of facioscapulohumeral dystrophy. Neurology 2014;
83:1056-1059.
7. Fanin M, Nascimbeni AC, Fulizio L, Angelini C. The frequency of limb girdle muscular dystrophy 2A in northeastern Italy.
Neuromuscul Disord 2005;15:218-224.
8. Poppe M, Cree L, Bourke J, Eagle M, Anderson L, Birchall D, et al. The phenotype of limb-girdle muscular dystrophy type 2I.
Neurology 2003;60:1246-1251.
9. Sveen ML, Schwartz M, Vissing J. High prevalence and pheno- type–genotype correlations of limb girdle muscular dystrophy type 2I in Denmark. Ann Neurol 2006;59:808-815.
10. North KN, Wang CH, Clarke N, Jungbluth H, Vainzof M, Dowling JJ, et al. Approach to the diagnosis of congenital myopathies. Neuromuscul Disord 2014;24:97-116.
11. Nowak KJ, Wattanasirichaigoon D, Goebel HH, Wilce M, Pelin K, Donner K, et al. Mutations in the skeletal muscle α-actin gene in patients with actin myopathy and nemaline myopathy. Nat Genet 1999;23:208-212.
12. Goebel HH, Piirsoo A, Warlo I, Schofer O, Kehr S, Gaude M.
Infantile intranuclear rod myopathy. J Child Neurol 1997;12:
22-30.
13. Bornemann A, Petersen MB, Schmalbruch H. Fatal congenital
myopathy with actin filament deposits. Acta Neuropathol 1996;
92:104-108.
14. Laing NG, Clarke NF, Dye DE, Liyanage K, Walker KR, Kobayashi Y, et al. Actin mutations are one cause of congenital fi- bre type disproportion. Ann Neurol 2004;56:689-694.
15. Hung RM, Yoon G, Hawkins CE, Halliday W, Biggar D, Vajsar J.
Cap myopathy caused by a mutation of the skeletal alpha-actin gene ACTA1. Neuromuscul Disord 2010;20:238-240.
16. Zvaritch E, Kraeva N, Bombardier E, McCloy RA, Depreux F, Holmyard D, et al. Ca2+ dysregulation in Ryr1I4895T/wt mice causes congenital myopathy with progressive formation of mini- cores, cores, and nemaline rods. Proc Natl Acad Sci U S A 2009;
106:21813-21818.
17. Wallgren‐Pettersson C, Sainio K, Salmi T. Electromyography in congenital nemaline myopathy. Muscle Nerve 1989;12:587-593.