Ⅰ. 서 론
Apert syndrome은 관상봉합의 조기유합에 의한 첨두증 (acrocephaly)과 합지증(syndactyly)이 함께 나타나는 선천성 유전질환이다. 1906년 프랑스의 소아과 의사인 Eugene Apert 에 의해 처음 서술되었으며, 약 65,000~160,000당 1명꼴로 발생되는 것으로 알려져 있다1,2). 두개골 조기유합증 (Craniosynostosis)의 한 형태로서 상염색체 우성 유전(auto- somal dominant)을 하며, FGFR2(fibroblast growth factor receptor 2)의 점 돌연변이로 인해 일어난다1,3-5). 두개골의 봉 합은 조기에 유합되고, 두부는 비대칭적인 성장과 형태의 기형 을 보이게 된다6).
임상적으로 특징적인 손과 발의 합지증을 보이는 것 이외에
도 높은 이마와 편평한 뒤통수, 후퇴된 중안면부를 보이며, 안 구의 돌출이 관찰되고 양안 격리증과 밑으로 쳐진 안검열을 보 인다. 또한 시력의 이상이 관찰되는데, 시력의 이상은 돌출된 안구의 만성적인 노출, 두개내압의 증가, 시신경의 압박 등에 의해 일어날 수 있다. 중안면부는 두드러지게 후퇴되고, 상악골 은 저형성되어 있으며, 이로 인하여 하악골이 상대적으로 전돌 된 양상을 보이게 된다1,7). 코는 콧등의 함몰과 함께 짧고 넓은 형태를 보이고, 입술은 전형적으로 사다리꼴 모양을 나타내며, 하순은 돌출된 듯한 모습을 보인다1,6,8). 감소된 비인두와 좁아진 후비공은 어린 아이에서 호흡장애를 일으킬 수 있으며, 이러한 것에 대한 보상작용으로서 대부분의 유아는 구호흡을 하게 되 고, 개방교합을 초래하게 된다1). 이러한 호흡장애는 수면 중 무 호흡증을 유발할 수도 있다1,9). 또한 유아는 개교로 인하여 수유
Apert syndrome : 증례보고
박광선∙박호원∙이주현∙서현우
강릉대학교 치과대학 소아치과학교실 및 구강과학연구소
Apert syndrome은 관상봉합의 조기유합에 의한 첨두증(acrocephaly)과 합지증(syndactyly)이 함께 나타나는 선천성 유 전 질환으로서, 1906년 Apert에 의해 보고된 증후군이다. 약 65,000~160,000 당 1명꼴로 발생되며, FGFR2(fibroblast growth factor receptor 2)의 돌연변이로 생겨난다고 알려져 있고, 상염색체 우성 유전을 한다.
임상적으로 첨단두증(acrobrachycephaly)을 보이고, 특징적으로 손과 발의 합지증을 보이며, 높은 이마와 평평한 뒤통수, 후퇴된 중안면부, 안구돌출, 시력이상, 양안격리증, 밑으로 쳐진 안검열, 상악골 형성부전, 상대적인 하악 전돌의 양상 등을 보인다. 감소된 비강인두와 좁아진 후비공으로 인한 구호흡과 전방부 개교를 보이며, 이완시에 입술의 모양은 사다리꼴 형태 를 보인다. 중이염이 흔하고, 청각장애를 유발하기도 하며, 정신지체가 높은 비율로 나타난다. 특징적인 구내소견으로 연구개 열 또는 구개수열과 가성 경구개열, V자 모양의 악궁과 치열의 총생 등이 관찰되며, 전치부 개방교합과 구치부 교차교합을 동 반한 Class III 부정교합을 보인다.
본 증례는 강릉대학교 치과병원 소아치과에 내원한 6세 3개월의 남아로 충치 치료를 받고 싶다는 것을 주소로 내원하였다.
이 환자에서 보이는 Apert syndrome의 특징적인 치과적 소견에 대해 보고하는 바이다.
주요어 : Apert, 조기유합증, 합지증, 첨두증 국문초록
교신저자 : 박 호 원
강원도 강릉시 지변동 123 번지 / 강릉대학교 치과대학 소아청소년치과학교실 / Tel: 033-640-3157 / E-mail: [email protected]
와 발음시 기능적 장애를 보일 수 있다10).
상기도와 하기도의 비정상적인 형태 이외에도 청력에도 영향 을 줄 수 있으며, 중이염이 흔하고, 청각장애를 유발하기도 한 다1). 피부 증상으로서 여드름(Acne), 다한증(Hyperhidrosis), 저색소침착증(Hypopigmentation), 과각화증(Hyper- keratosis) 등이 관찰되며 여드름은 전형적으로 심하고, 소년기 또는 조기 사춘기에 발생하며, 팔 등 다양한 부위에 이환된
다1,11,12). 이밖에도 심장혈관계(Cardiovascular), 비뇨생식계
(Genitourinary), 위장관계(Gastrointestinal)의 이상도 보고 되었다13). 정신 지체는 높은 비율로 나타나지만, 지능이 정상인 경우도 있다1,14).
특징적인 구내소견으로 연구개열(cleft of the soft palate) 또는 구개수열(bifid uvula)과 가성 경구개열(pseudocleft of the hard palate)을 보이며, 상악골의 저형성으로 인한 V자 모 양의 악궁과 치열의 총생이 관찰되고, 전치부 개방교합과 구치 부 교차교합을 동반한 Class III 부정교합을 보인다1). 또 다른 구내소견으로 치열의 매복, 맹출지연 등이 관찰되며, 때때로 과 잉치, 선천성 결손 등이 관찰될 수 있다1,8). 치아의 형태 이상으 로서, Shovel shaped incisors 가 환자의 1/3에서 보고되었다
1,6,7,11).
본 증례는 강릉대학교 치과병원 소아치과에 내원한 6세 3개 월의 남아로 이미 타 병원(아산재단 강릉병원)에서 Apert syn- drome의 진단을 받고, 성형외과, 신경외과, 소아과, 정형외과 에서 두개골 유합과 합지증에 대한 수술을 한 상태였으며, 치아 우식증 치료를 위해 본원 소아치과로 의뢰되었다. 이 환자에서 Apert syndrome의 특징적인 치과적 소견을 발견할 수 있었기 에 보고하는 바이다.
Ⅱ. 증 례
6세 3개월된 남자 아이로 2006년 6월 5일 충치치료를 주소 로 내원하였다(Fig. 1). 과거력을 보면, 2000년 3월 제왕절개 술을 통해 분만하였고, 출생시의 체중은 3.2kg 였다. 출생시 얼 굴의 기형, 두개골 조기 유합, 커져있는 천문, 튀어나온 이마, 양쪽 손과 발에 합지증 등 복합적인 기형을 보였고, Apert syndrome으로 진단받았다. 가족력은 없었고, 출생시 아버지의 나이는 42세, 어머니는 32세 였으며, 다른 형제는 없다.
생후 4개월에 두개 절제술로서 Frontal advancement op- eration을 시행하여, 전두골과 안와를 전진시키고, 두개의 용적 을 넓혔다. 6세경에는 합지증해결을 위한 수술을 시행하였고, 2007년 12월 추가적인 손가락 수술이 시행될 예정이다. 발가 락 수술은 아직 시행되지 않았으며, 성장이 더 진행된 후 시행 될 예정이다.
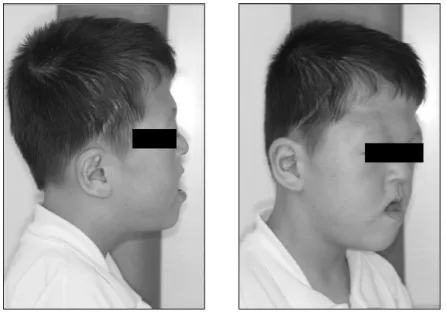
임상적 소견으로서 돌출된 안구와 양안격리증이 관찰되었으 며, 밑으로 처진 안검열을 보였다(Fig. 2). 이마는 튀어나와 있 었으며, 뒤통수는 편평하였다. 중안면부는 성장 부족으로 인하 여 후퇴되어 있었고, 상악골의 저성장과 함께 하악골의 상대적 전돌의 양상이 관찰되었다. 하순은 돌출된 듯한 모습을 보이고 있었고, 입은 사다리꼴 모양으로 벌어져서 전형적인 Apert syndrome 환자의 모습을 보이고 있었다(Fig. 3). 코는 짧고 넓은 형태를 보였고, 콧등은 함몰되어 있었다(Fig. 2, 3).
측방 두부규격 방사선사진에서도 상악골 형성부전, 상대적인 하악 전돌 양상을 관찰할 수 있었으며, 이와 함께 심한 개교와 전치부 반대교합을 보이고 있었다. 또한 이전의 두개골 성형술 시 부착된 금속판과 강선결찰이 관찰되고, 두개내압 상승으로 인한 두개부의 지압흔(digital impression)을 보였다(Fig. 4).
손과 발의 합지증이 관찰되었고(Fig. 5), 손은 합지증에 대한 수술을 시행한 상태로 내원하였으며, 추후 추가적인 수술이 시 행될 계획이다(Fig. 6). 시력과 청력에는 이상이 없었으며, 정 신지체가 있었다.
Fig. 1. Extraoral facial view. Fig. 2. Extraoral facial view.
Fig. 3. Extraoral facial view.
Fig. 4. Lateral skull projection (arrow:digital impression).
임상검사 시 구내 소견으로서, 상악 전치부, 상악 구치부, 하 악 구치부(#51, 52, 53, 55, 61, 62, 65, 74, 85)에 치아우식 증이 관찰되었고, 하악 제2유구치는 amalgam 으로 수복되어
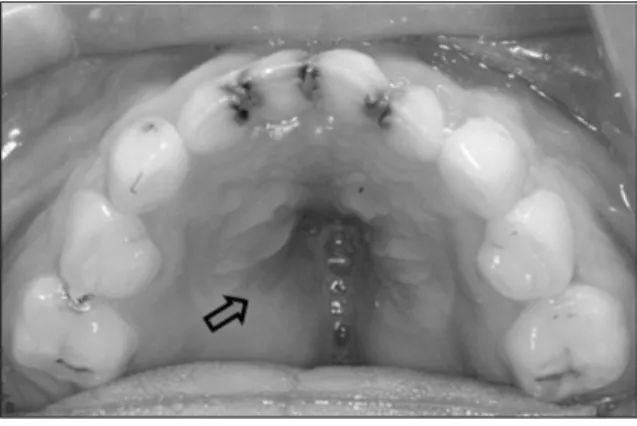
있는 상태였다. 하악 유측절치는 자연탈락 상태였으며, 하악 제 1대구치와 하악 중절치가 맹출중이었고, 상악 제1대구치는 아 직 맹출하지 않았다. 전방부에서 개교가 관찰되었으며, 구개부 에 연구개열은 존재하지 않았고, 경구개의 외측으로 구개부 조 직의 양측성 비대로 인한 가성 경구개열이 관찰되었다. 상악궁 과 하악궁의 협착으로 인한 V자 모양의 좁은 악궁이 존재하였 고, 상악 구치와 하악 구치의 설측 경사를 동반한 구치부 교차 교합이 관찰되었다. 전치부에서는 큰 음의 값의 수평피개를 보 였다(Fig. 7, 8, 9, 10).
치아우식증에 대한 치료로서 상악 좌측 제2유구치, 상악 우 측 제2유구치를 물리적 속박하에 2006년 6월과 7월, 두 차례 에 걸쳐 아말감 수복을 하였다. 다른 치아우식증 부위는 정지된 우식 병소 또는 초기 우식 병소로 판단되어 정기적인 검진 후 필요시에 치료하기로 하였다. 치료 후 3개월 정기 검진시에 파 노라마 방사선 사진에서 과잉치, 선천성 결손 등 치아 수의 이 상은 발견되지 않았고, 상악궁에 치아 맹출 공간의 부족이 예상 되었다(Fig. 11).
Fig. 7. Upper occlusal view (arrow: Pseudocleft of the hard palate).
Fig. 8. Frontal view.
Fig. 9. Overjet and Overbite.
Fig. 11. Panoramic view.
Fig. 10. Lower occlusal view.
Ⅲ. 총괄 및 고찰
Apert syndrome은 FGFR2(fibroblast growth factor re- ceptor 2)의 돌연변이로 일어나는 두개골 조기유합증 (Craniosynostosis)의 한 형태이다. 두개골 조기유합증은 두개 봉 합 의 조 기 유 합 으 로 정 의 되 며 , Crouzon, Apert, Carpenter, Chotzen, Pfeiffer syndrome 등으로 나누어 살펴 볼 수 있다. Apert syndrome은 모든 두개골 조기유합증의 4.5%에 해당하며2,15), 특징적인 손과 발의 합지증으로 감별진단 이 가능하다. 주로 제 2, 3, 4지의 융합의 관찰되고, 엄지와 제 5지의 융합도 관찰될 수 있다16).
두개골의 기형은 두개골의 조기 융합에 의해 일어나는데, 특 히 관상봉합 (coronal suture)의 조기융합으로 인해 두개골의 모양은 수직으로 길어져 높으며, 그 정점은 대, 소천문 사이에 위치한다. 안면은 뚜렷하게 넓고, 두부는 전후경이 짧으며 후두 부가 납작하고 전두부는 돌출한다. 두개골의 조기융합으로 인 하여 뇌실질 조직의 성장에 따라 뇌척수액압이 증가하게 되는 데 이로 인해 시력장애나 지능저하를 초래할 수 있다17). 두부 방 사선 사진에서 두개강 내압의 증가로 인해, Crouzon syn- drome과 유사한 지압흔(digital impression)이 관찰될 수 있 으며, 두개궁의 내측 부위가 움푹 들어간 모양의 방사선 투과성 부위로서 다발성으로 나타난다1,18). 본 환자의 측방 두부규격 방 사선 사진에서도 두개골 성형술시 부착된 금속결찰과 두개내압 상승으로 인한 지압흔을 관찰할 수 있었다(Fig. 4).
Apert syndrome은 상 염 색 체 우 성 으 로 유 전 되 며 , FGFR2(fibroblast growth factor receptor 2)에서의 유전자 돌연변이에 의해 발생되는데 대부분 Ser252Trp(S252W), 또 는 Pro253Arg(P253R)의 점 돌연변이로 인하여 일어난다1,3-5).
Apert syndrome에서 발생빈도와 부모의 연령 사이에 상관 성이 보고되었는데, Moloney 등19)은 부모의 나이가 35세 이상 일 때 더 높은 빈도를 나타냄을 보고하였고, 특히 아버지의 연 령 증가는 새로운 돌연변이의 가능성을 높여, 발생빈도와 상관 성을 보인다고 하였다. 본 환자의 경우 출생시 아버지의 나이는 42세, 어머니는 32세로서 양친의 나이가 많음을 보였고, 특히
아버지의 나이가 많은 것을 관찰할 수 있었다.
본 증후군은 다방면의 협진에 의해서 심미적, 기능적 결손을 치료해 줄 수 있는데, 두개골의 조기 융합전에 신경외과적 처 치, 안면기형 및 기능적 교정, 양안격리증의 치유, 수부 및 족부 의 기형 교정을 치료의 목표로 한다20).
두부에 대한 신경외과적 치료는 시력감소와 정신발육 부전의 치료 및 예방에 목적을 두며, 특히 정상적 두뇌의 성장은 생후 6개월이면 출생시 무게의 2배가 될 정도로 빠르고, 뇌압상승이 생후 1년 이내에 가장 현저하므로 1세 이전의 조기 두개 성형 술이 권장되고 있다17,21). 전통적으로 Fronto-orbital advance- ment를 4~9개월 경에 시행하며, 이들 환자중 약 75%는 2세 경에 Second Fronto-orbital advancement를 시행하게 된다22). 본 환자의 경우 생후 4개월에 두개 절제술로서 Frontal ad- vancement operation을 시행하였고, 이로서 전두골과 안와를 전진시키고, 두개의 용적을 넓혔다.
Apert syndrome, Crouzon syndrome, Hemifacial micro- somia 등 발육부전을 포함한 일부의 선천적인 문제들을 해결하 기 위해서는 유아기 또는 유년기 초기의 수술을 필요로 한다23). 대부분은 상악골 저형성으로 인한 Class III 부정교합을 보이므 로, 교정적 치료와 외과적 술식을 병행하여 진행하게 되는데, 악교정술은 저성장된 상악을 하악과 조화시키기위해 시행하게 되며, 성장과 발육이 왕성한 시기에 이를 이용하여 저성장된 상 악골의 전방성장을 도모하고, 좁은 상악궁에 대한 치료를 시행 하게 된다. 외과적 술식으로 중안면부를 전진시켜서 상악골과 하악골의 골격적 관계를 바로 잡아주고, 교합관계를 개선시킬 수 있으며, 상기도를 통한 호흡 기능의 개선이 이루어 질 수 있 고, Nasolabial angle의 변화에 따른 Nasal airway patency 가 개선되어 비호흡의 개선도 이루어질 수 있다. 또한 돌출된 안구 보호의 효과도 얻을 수 있다22,24).
Hohoff 등10)은 Midface advancement는 거의 항상 필수적 이며, 보통 Cranial surgery의 반복이 필요하다고 하였으며, 문헌에서 독일 Mu¨nster 대학병원의 치료개념을 소개하였다 (Table 1).
Table 1. Interdisciplinary treatment concept for patients with Apert syndrome at the University Hospital of Mu¨- nster(individual adaptions allowed)
Stage Age Procedure
1 3-6 months Eradicative-osteoclastic method
2 7-10 years LeFort III osteotomy+
Frontal advancement+
Protraction face mask immediately postoperatively for 6-12 months(rapid stenosis, problems with anchage of the orthodontic appliance due to exfolia- tion of deciduous teeth, and eruption of permanent teeth)
Early LeFort III osteotomy might help to avoid a (second) LeFort III osteoto- my at stage 5, and thus reduce surgical risks.
3 > 9 years Extraction therapy
Bruce25)에 의하면 Midfacial surgery는 가능한 한 늦추는 것이 바람직하지만, 많은 경우에서 4~6세 경에 수술이 필요하 다. 이러한 Early midface advancement는 해부학적, 심리학 적으로 이득을 줄 수 있고 수면 무호흡증의 감소등 기능적인 이 득도 얻을 수 있지만, 이른 시기에 수술을 할수록 추후에 추가 적인 수술을 하게 될 가능성은 커진다. Meazzini 등26)은 Early childhood 시기(평균: 7.3세, 범위: 4.8~10세)에 LeFort III advancement를 시행한 환자들의 두부계측방사선사진 연구에 서 술후 상악 분절의 안정성이 두드러짐을 보고하였으나, 일반 적으로 성장에 결정적인 효과나 이득을 주지 못하므로 유년기 에 필수적인 수술은 아니라고 하였다.
최근에 사용되는 RED(rigid external distraction) system 등을 이용한 Distraction Osteogenesis의 장점은 심하게 저형 성된 상악을 재위치시키고 유지하는데 Bone graft 또는 Rigid fixation이 필요없다는 것이고, 이전의 Internal device가 지녔 던 Vector control의 한계를 극복할 수 있었으며 골격적 성숙이 완성될 때까지 기다릴 필요가 없어서 심각한 중안면부 저형성 환자의 기능적, 심미적, 심리적 한계를 치료할 수 있다는 것이 다. 또 다른 장점으로 Distraction osteogenesis를 시행함으로 서 연조직 신장(muscles, nerves, vessels, mucosa, skin 등) 의 효과로 상악골을 전통적인 Osteotomy 보다 더 많이 Advancement 시킬 수 있게 되었다24). 이러한 치료는 4세 이상 의 상악골 저형성증 환자에게 매우 효과적이고, 예측 가능하고 안정적이다. 하지만, 성숙이 완료된 후 2차“finishing”수술의 가능성은 남아있다22).
Apert syndrome의 합지증은 이완술을 시행하여, 수부의 기 능항진을 유도하여 함으로써 해결할 수 있다. Bauer 등27)은 융 합된 지골을 조기 분리 시켜야만 수지골의 정상발육과 연부 조 직 및 피부의 성장이 조화를 이룬다고 기술하였고, Hoover28)는 합지증 이완술을 생후 2년 내에 시행하는 것이 예후가 좋다고 보고하였으나, 수술 후 환자가 의사 및 물리치료사의 치료에 협 조할 수 있는 연령이 되어야 한다고 하였다. 본 환자는 6세경에 합지증 이완술을 시행한 상태였고, 이후 추가적인 손가락 수술 이 시행될 예정이었다. 발가락 수술은 아직 시행되지 않은 상태 였으며, 성장과 협조도를 고려하여 추후 시행될 예정이다.
전형적인 치아와 구강내 소견은 아주 높은 빈도로 발견된다.
상악골은 저형성으로 인한 V자 모양의 악궁을 보이며, 치열은 총생을 보이고, 전치부 개방교합과 구치부 교차교합을 동반한 Class III 부정교합을 보인다1). 또한 특징적으로 연구개열 또는 구개수열과 가성 경구개열을 보이는데, 구개부 치은의 비대는 주로 Hyaluronic acid로 구성된 Mucopolysaccharides의 축 적으로 생기며, 경구개의 외측을 따라서 관찰된다1,29). 이러한 치은의 비대는 종종 나이가 들면서 커지고 경구개의 Pseudocleft를 보이게 된다1). Mucopolysaccharides의 축적은 하악 치은에서도 관찰될 수 있으나, 그 정도는 상악에 비해 덜 하다30). 진성 경구개열은 증례의 약 23.5%에서 관찰되며, 연구 개열 또는 구개수열은 모든 환자의 75%에서 관찰된다1,7,31). 본
환자의 경우에서도 상악골 협착을 동반한 가성 경구개열을 보 였다. 연구개열과 구개수열은 관찰되지 않았다.
Apert syndrome은 매복, 총생, 맹출지연을 보일 수 있고, 때때로 과잉치 또는 선천적 결손 등이 관찰되기도 한다.
Kaloust 등30)은 Apert syndrome 환자의 치열 발육이 평균 0.96년(0.5~2.9년) 지연된다고 하였고, 그 정도는 연령의 증 가에 비례하여 심해진다고 하였는데, 7세 이하는 평균 0.23년, 7세에서 10세는 평균 0.97년, 11세 이상은 평균 1.7년 정도가 지연된다고 하였다. 또한 치은의 비대가 치아의 맹출지연과 관 련이 있어 보인다는 의견이 있으나, Kaloust 등30)은 치아맹출 의 지연은 다른 국소 요인의 작용으로 보이며, Muco- polysaccharides의 축적이 발육지연의 필수적인 요소는 아니 라고 하였다1). 치아의 맹출지연은 이차적으로 악골내에서 치아 의 총생과 변위를 야기할 수 있으며, 종종 치배가 두 열로 넓게 배열되는 경우도 있다8). 치아의 형태이상으로서 Shovel shaped incisors가 보고되었으며, Letra 등32)은 환자의 17명 의 환자 중 7명(41%)에서 치아의 선천적 결손을 보고하였다
1,6,7,11). 본 환자에서는 치아의 형태이상이나 치아 개수의 이상은
발견되지 않았으나, 절치가 맹출하기 전이므로 맹출 후 지속적 인 관찰이 필요할 것으로 생각된다.
Apert syndrome 환자의 구강위생관리는 매우 중요하지만, 어려운 부분 중 하나이다. 손의 형태이상은 치실질과 잇솔질을 어렵게 만든다. Floss holder 나 전동칫솔, 불소양치액등으로 구강위생관리를 양호하게 하도록 도울 수 있다. 전문가 관리로 서 주기적인 검진을 자주 시행해 주어야 하며, 치면세마, 불소 도포, 치면 열구 전색등이 강조될 수 있다33). 본 환자의 경우 현 재 주기적인 검진을 통해 치면세마를 시행하고 있으며 추후 필요시 전문가 불소 도포 또는 치면 열구 전색을 시행할 예정 이다.
향후 치료계획으로서 저성장된 상악골을 LeFort III os- teotomy를 동반한 Distraction osteogenesis 등으로 최대한 악골과 치열의 부조화를 작게 한 후, 향후 재평가를 통해 교정 치료를 동반한 외과적 수술로 더 조화로운 안모와 향상된 교합 을 유도하는 것이 바람직할 것으로 생각된다. 하지만 본 환자는 정신지체를 지니고 있어 협조가 어려웠고, 타과의 잦은 수술과 내원으로 인한 피로와 경제적 사정으로 인하여 보호자의 협조 를 얻기 힘들었다. 현재는 수복 치료를 시행한 상태이며, 향후 협조를 얻어 계획된 치료를 진행하는 것이 필요하다.
Ⅳ. 요 약
강릉대학교 치과병원 소아치과에 내원한 Apert syndrome 환자로서 특징적인 치과적 소견을 발견할 수 있었고, 임상검사 를 통해서 다음과 같은 지견을 얻을 수 있었다.
1. 전형적인 임상적 소견으로서 돌출된 안구와 양안격리증이 관찰되었으며, 밑으로 처진 안검열을 보였다.
2. 튀어나온 이마와 평평한 뒤통수, 후퇴된 중안면부를 보였
고, 하악골은 상대적으로 전돌된 양상을 보이고 있었다.
3. 손과 발의 합지증을 확인할 수 있었다.
4. 하순은 돌출된 듯한 모습을 보였고, 입은 전형적인 사다리 꼴 모양을 보이고 있었다.
5. 구강내 소견으로 전방부 개교, 가성 경구개열, V자 모양의 좁은 악궁을 관찰할 수 있었고, 전치부와 구치부에서 반대 교합이 관찰되었다.
6. 향후 치료계획으로서 정기검진을 통한 구강위생관리를 시 행할 예정이며, 향후 재평가를 통해 외과적 수술을 동반한 교정치료로서 안모의 개선과 교합의 향상을 유도해야 할 것으로 생각된다.
참고문헌
1. Brad W. Neville, Douglas D. Damm, Carl M. Allen, et al. : Oral & Maxillofacial Pathology second edi- tion, Saunders, Philadelphia, 40-42, 2002.
2. Cohen MM Jr, Kreiborg S, Lammer EJ, et al. : Birth prevalence study of the Apert syndrome. Am J Med Genet, 42:655-659, 1992.
3. Fanganiello RD, Sertie′AL, Reis EM, et al. : Apert p.Ser252Trp mutation in FGFR2 alters osteogenic potential and gene expression of cranial periosteal cells. Mol Med, 13:422-442, 2007.
4. Jadico SK, Young DA, Huebner A, et al. : Ocular abnormalities in Apert syndrome: genotype/pheno- type correlations with fibroblast growth factor recep- tor type 2 mutations. J AAPOS, 10:521-527, 2006.
5. Wilkie AO, Slaney SF, Oldridge M, et al. : Apert syndrome results from localized mutations of FGFR2 and is allelic with Crouzon syndrome. Nat Genet, 9:165-172, 1995.
6. Cohen MM Jr, Kreiborg S : A clinical study of the craniofacial features in Apert syndrome. Int J Oral Maxillofac Surg, 25:45-53, 1996.
7. Kreiborg S, Cohen MM Jr : The oral manifestations of Apert syndrome. J Craniofac Genet Dev Biol, 12:41-48, 1992.
8. Ferraro NF : Dental, orthodontic, and oral/maxillo- facial evaluation and treatment in Apert syndrome.
Clin Plast Surg, 18:291-307, 1991.
9. Lo LJ, Chen YR : Airway obstruction in severe syn- dromic craniosynostosis. Ann Plast Surg, 43:258- 264, 1999.
10. Hohoff A, Joos U, Meyer U, et al. : The spectrum of Apert syndrome: phenotype, particularities in ortho-
surgery. Head Face Med, 3:10, 2007.
11. DeGiovanni CV, Jong C, Woollons A : What syn- drome is this? Apert syndrome. Pediatr Dermatol.
24:186-188, 2007.
12. Cohen MM Jr, Kreiborg S : Cutaneous manifesta- tions of Apert syndrome. Am J Med Genet, 58:94- 96, 1995.
13. Cohen MM Jr, Kreiborg S : Visceral anomalies in the Apert syndrome. Am J Med Genet, 45:758-760, 1993.
14. Cohen MM Jr, Kreiborg S : Hands and feet in the Apert syndrome. Am J Med Genet, 57:82-96, 1995.
15. Park WJ, Theda C, Maestri NE, et al. : Analysis of phenotypic features and FGFR2 mutations in Apert syndrome. Am J Hum Genet, 57:321-328, 1995.
16. Richard E. Behrman, Robert M. Kliegman, Hal B.
Jenson. : Nelson Textbook of Pediatrics 17th edi- tion, Saunders, Philadelphia, 1992-1993, 2004.
17. 임창규, 박상철 : Apert 증후군(Acrocephalosyndactyly) 1례. The journal of Soonchunhyang University, 15:679-687, 1992.
18. 대한구강악안면방사선학교수협의회: 구강악안면방사선학 제3판, 나래출판사, 562-563, 2001.
19. Moloney DM, Slaney SF, Oldridge M, et al. : Exclusive paternal origin of new mutations in Apert syndrome. Nat Genet, 13:48-53, 1996.
20. 황준경, 이동식, 임정근 등: Apert syndrome(1례보고), 대한정형외과학회지, 21:939-942, 1986.
21. 임용걸, 이은주, 옥광익 등 : Apert 증후군 환자의 마취 경 험 1례(증례보고). 대한마취과학회지, 29:300-303, 1995.
22. Mikhail L. Samchukov, Jason B. Cope, Alexander M. Cherkashin : Craniofacial Distraction Osteogenesis, Mosby, Texas, 477-544, 2002.
23. 양원식, 김태우, 백승학 : 최신치과교정진단학, 지성출판 사, 서울, 325-334, 2007.
24. Anderson PJ, Tan E, David DJ : Simultaneous mul- tiple vector distraction for craniosynostosis syn- dromes. Br J Plast Surg, 58:626-631, 2005.
25. Bruce DA. : Consensus: craniofacial synostoses.
Apert and Crouzon syndromes. Childs Nerv Syst, 12:734-736, 1996.
26. Meazzini MC, Mazzoleni F, Caronni E, et al. : Le Fort III advancement osteotomy in the growing child affected by Crouzon's and Apert's syndromes:
presurgical and postsurgical growth. J Craniofac
27. Bauer TB, Tendra JM, Trusler HM : Technical mod- ification in repair of syndactylism. Plast. and Reconst. Surg, 17:385-392, 1956.
28. Hoover GH, Flatt AE, Weiss MW : The hand and Apert's syndrome. J Bone Joint Surg Am, 52:878- 895, 1970.
29. Solomon LM, Medenica M, Pruzansky S, et al. : Apert syndrome and palatal mucopolysaccharides.
Teratology, 8:287-291, 1973.
30. Kaloust S, Ishii K, Vargervik K : Dental develop- ment in Apert syndrome. Cleft Palate Craniofac J, 34:117-121, 1997.
31. Arroyo Carrera I, Marti′nez-Fri′as ML, Marco Pe′rez JJ, et al. : Apert syndrome: clinico-epidemiological analysis of a series of consecutive cases in Spain. An Esp Pediatr, 51:667-672, 1999.
32. Letra A, de Almeida AL, Kaizer R, et al. : Intraoral features of Apert's syndrome. Oral Surg Oral Med Oral Pathol Oral Radiol Endod, 103:38-41, 2007.
33. Tosun G, Sener Y : Apert syndrome with glucose-6- phosphate dehydrogenase deficiency: a case report.
Int J Paediatr Dent, 16:218-221, 2006.
34. 송수복, 김정욱, 김종철 : Apert syndrome 환자의 제증상 에 관한 증례보고. 대한소아치과학회지, 29:44-50, 2002.
35. 손흥규, 김순주, 최병재 등 : Crouzon's disease 의 증례.
대한소아치과학회지, 11:249-254, 1984.
36. 양규호, 오희수 : 상악골 저성장을 동반한 Crouzon syn-
drome에 관한 증례. 대한소아치과학회지, 18:106-116, 1991.
37. Slaney SF, Oldridge M, Hurst JA, et al. : Differential effects of FGFR2 mutations on syn- dactyly and cleft palate in Apert syndrome. Am J Hum Genet, 58:923-932, 1996.
38. Jadico SK, Young DA, Huebner A, et al. : Ocular abnormalities in Apert syndrome: genotype/pheno- type correlations with fibroblast growth factor recep- tor type 2 mutations. J AAPOS, 10:521-527, 2006.
39. Tolarova MM, Harris JA, Ordway DE, et al. : Birth prevalence, mutation rate, sex ratio, parents' age, and ethnicity in Apert syndrome. Am J Med Genet, 72:394-398, 1997.
40. Shetye PR, Boutros S, Grayson BH, et al. : Midterm follow-up of midface distraction for syndromic cran- iosynostosis: a clinical and cephalometric study.
Plast Reconstr Surg, 120:1621-1632, 2007.
41. Blank CE : Apert's syndrome (a type of acro- cephalosyndactyly) - observations on a British series of thirty-nine cases. Ann Hum Genet, 24:151-164, 1960.
42. Marchac D, Renier D, Broumand S : Timing of treatment for craniosynostosis and facio-craniosyn- ostosis: a 20-year experience. Br J Plast Surg, 47:211-222, 1994.
Abstract
APERT SYNDROME : CASE REPORT
Kwang-Sun Park, Ho-Won Park, Ju-Hyun Lee, Hyun-Woo Seo
Department of Pediatric Dentistry, Oral Science Research Center, College of Dentistry, Kangnung National University
Apert syndrome is an autosomal dominant condition characterized by craniosynostosis, midface hypoplasia, and syndactyly of the hands and feet. It occurs in about 1 of every 65,000 to 160,000 births and is caused by a mutation in the fibroblast growth factor receptor 2(FGFR2) gene.
Apert syndrome typically produces acrobrachycephaly(tower skull). The occiput is flattened, and there is a tall appearance to the fore head. Ocular proptosis is a characteristic finding, along with hypertelorism and down- ward slanting lateral palpebral fissures. The middle third of the face is markedly retruded and hypoplastic, re- sulting in a relative mandibular prognathism. The reduced size of the nasopharynx and narrowing of the posteri- or choana can lead to mouth breathing, contributing to an open-mouth apprance. Three fourths of all patients exhibit either a cleft of the soft palate or a bifid uvula. The maxillary hypoplasia leads to a V-shaped arch and crowding of the teeth.
A 6-year-old male patient visited to the Department of Pediatric dentistry, Kangnung National University of Dental Hospital. He visited the hospital to get treatment of carious teeth.
The purpose of this report is to present a specific dental manifestations about the apert syndrome.