저작자표시-비영리-변경금지 2.0 대한민국 이용자는 아래의 조건을 따르는 경우에 한하여 자유롭게 l 이 저작물을 복제, 배포, 전송, 전시, 공연 및 방송할 수 있습니다. 다음과 같은 조건을 따라야 합니다: l 귀하는, 이 저작물의 재이용이나 배포의 경우, 이 저작물에 적용된 이용허락조건 을 명확하게 나타내어야 합니다. l 저작권자로부터 별도의 허가를 받으면 이러한 조건들은 적용되지 않습니다. 저작권법에 따른 이용자의 권리는 위의 내용에 의하여 영향을 받지 않습니다. 이것은 이용허락규약(Legal Code)을 이해하기 쉽게 요약한 것입니다. Disclaimer 저작자표시. 귀하는 원저작자를 표시하여야 합니다. 비영리. 귀하는 이 저작물을 영리 목적으로 이용할 수 없습니다. 변경금지. 귀하는 이 저작물을 개작, 변형 또는 가공할 수 없습니다.
Master’s Thesis in the Department of
Biomedical Science
Ivermectin kills cancer cells
via catastrophic changes
in the endoplasmic reticulum structure
Ajou University Graduate School
Major in Cancer Biology
Ivermectin kills cancer cells
via catastrophic changes
in the endoplasmic reticulum structure
Kyeong Sook Choi, Advisor
I submit this thesis as the
Master’s thesis in the Department of Biomedical Sciences
August 2019
Ajou University Graduate School
Major in Cancer Biology
i
-ABSTRACT-
Ivermectin kills cancer cells via catastrophic changes
in the endoplasmic reticulum structure
Ivermectin (IVM), an antiparasitic drug, is now considered a strong candidate for repurposing as an anticancer drug. IVM has been known to induce apoptosis or autophagy in several cancer cells, but the underlying mechanisms to explain its anticancer effects still remain unclear. In this study, we found that induces dramatic alterations of the endoplasmic reticulum (ER) structure, including reorganization, vacuolation, and subsequent permeabilization of the ER, prior to cancer cell death. In addition, IVM increases ER stress, and pretreatment with cycloheximide, a protein synthesis inhibitor, markedly blocked IVM-induced ER stress, vacuolation and permeabilization of the ER, and cell death, but not ER reorganization. Furthermore, IVM induces the activation of ERK and p38, imbalance of ions, including Ca2+ and Cl-, and inhibition of these signals effectively attenuated
IVM-induced vacuolation and permeabilization of the ER and subsequent cell death. Taken together, these results suggest that IVM-induced catastrophic changes in the ER, including the vacuolation and permeabilization of the ER, critically contribute to the anti-cancer effect of IVM. ER stress, imbalance of Ca2+ and Cl-, and activation of MAPK play crucial roles in this process.
ii
TABLE OF CONTENTS
ABSTRACT --- i
TABLE OF CONTENTS --- ii
LIST OF FIGURES --- iv
I.
INTRODUCTION --- 1
II.
MATERIALS AND METHODS--- 11
A. Chemicals and antibodies
--- 11
B. Cell culture
--- 12
C. Measurement of cellular viability
--- 12
D. Immunoblot analyses
--- 13
E. Morphological examination of the ER and mitochondria
--- 13
F. Immunofluorescence microscopy
--- 14
G. Transmission electron microscopy
--- 14
H. Measurement of intracellular Ca
2+levels
--- 14
I. Measurement of intracellular Cl
-levels
--- 15
J. Measurement of intracellular reactive oxygen species levels
--- 15
---iii
--- 15
L.
ATP measurement --- 16
M.
Statistical analysis --- 16
III.
RESULTS --- 17
1. Ivermectin (IVM) effectively induces cell death in various
cancer cells
--- 17
2. IVM induces catastrophic changes in the endoplasmic
reticulum structure
--- 23
3. ER reorganization is associated with lipid
--- 30
4. IVM induces paraptosis in MDA-MB 435S
--- 34
5. Increase in intracellular Ca
2+critically contributes to
IVM-induced paraptosis
--- 41
6. Imbalance in intracellular chloride may be involved in
IVM-induced paraptosis
--- 47
IV.
DISCUSSION --- 53
iv
LIST OF FIGURES
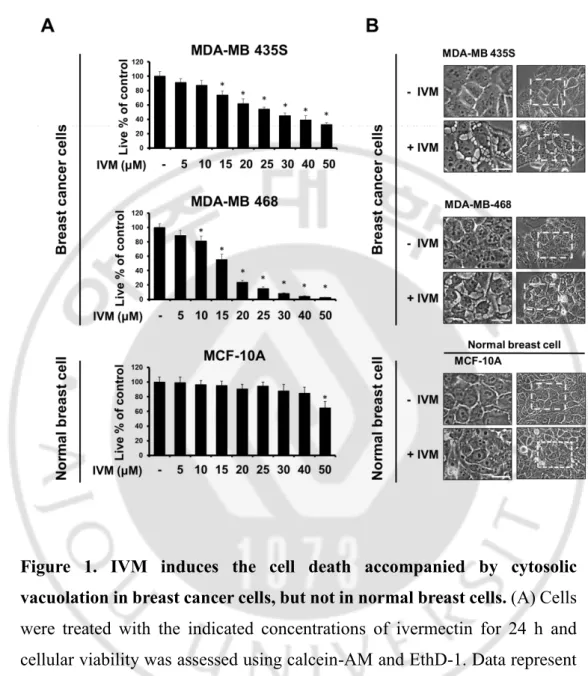
Figure 1. IVM induces the cell death accompanied by cytosolic
vacuolation in breast cancer cells, but not in normal breast cells
--- 19
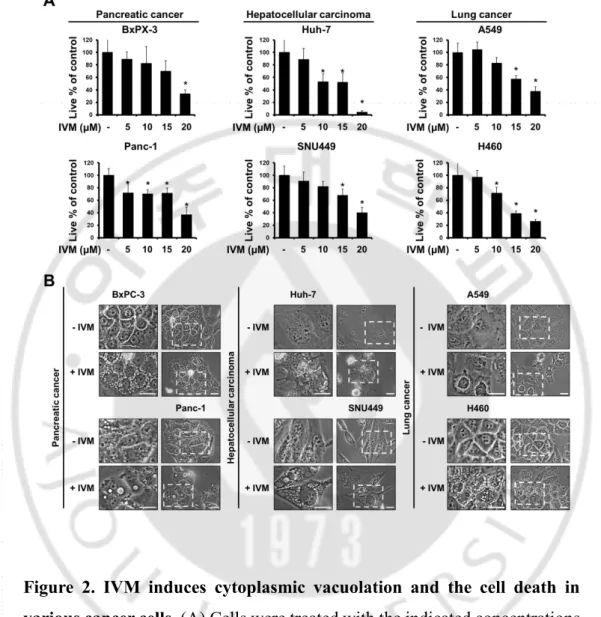
Figure 2. IVM induces cytoplasmic vacuolation and the cell death
in various cancer cells.
--- 20
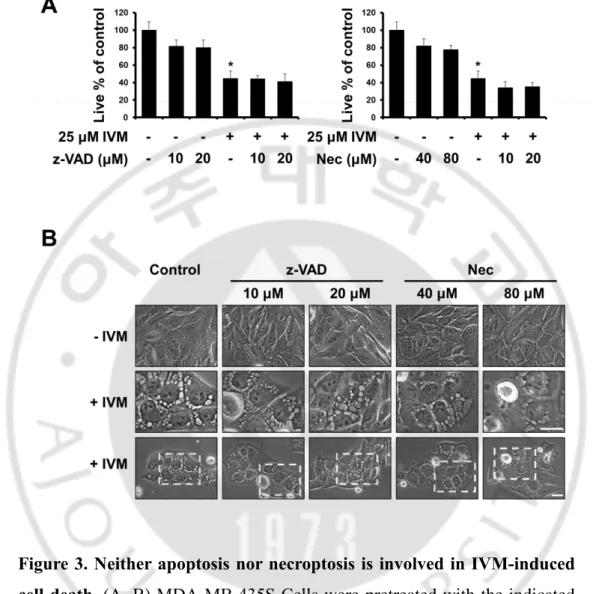
Figure 3. Neither apoptosis nor necroptosis is involved in
IVM-induced cell death
--- 21
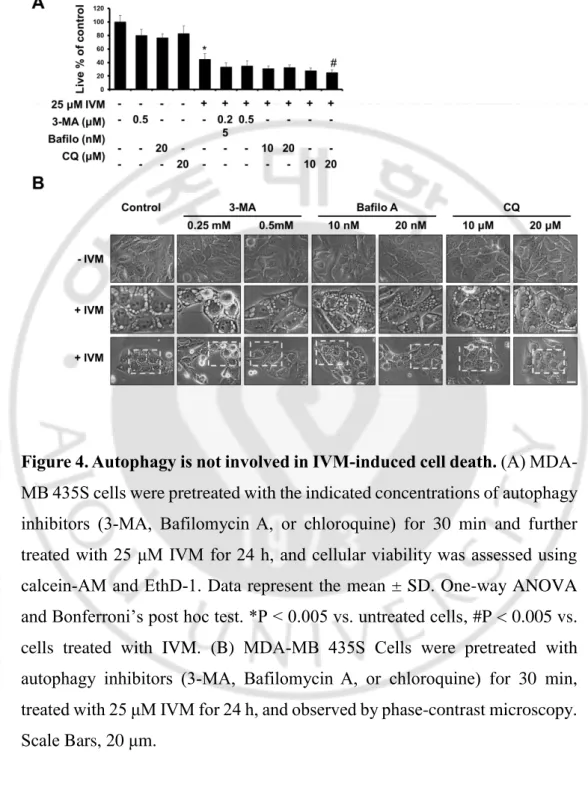
Figure 4. Autophagy is not involved in IVM-induced cell death
--- 22
Figure 5. IVM induces catastrophic changes in the ER in
YFP-ER/435S cells
--- 25
Figure 6. IVM induces catastrophic changes in the ER in GFP-
Sec61/435S cells
--- 26
Figure 7. IVM induces catastrophic changes in the ER in
MDA-MB 435S cells
--- 27
Figure 8. Electron microscopic observation of MDA-MB 435S
cells treated with IVM
--- 28
Figure 9. Time-lapse imaging of YFP-ER cells treated with
induced by IVM
--- 29
v
Figure 10. IVM induces lipid droplet
--- 31
Figure 11. Lipid is not associated with ER vacuolation and cell
death induced by IVM
--- 32
Figure 12. ER reorganization is associated the lipid
--- 33
Figure 13. IVM induces ER stress, which is blocked by CHX
--- 36
Figure 14. CHX blocks IVM-induced ER vacuolation and cell
death
--- 37
Figure 15. CHX blocks ER vacuolation and permeabilization but
not ER reorganization
--- 38
Figure 16. Activation of ERK and p38 significantly contributes to
IVM-induced vacuolation and cell death
--- 39
Figure 17. Inhibition of ERK and p38 delays IVM-induced ER
vacuolation and permeabilization
--- 40
Figure 18. IVM increases intracellular Ca
2+, which is inhibited by
BAPTA-AM
--- 43
Figure 19. BAPTA-AM blocks IVM-induced ER vacuolation and
cell death
--- 44
Figure 20. BAPTA-AM blocks IVM-induced ER vacuolation and
permeabilization
--- 45
Figure 21. IVM-induced Ca
2+increase is associated with ER stress
vi
--- 46
Figure 22. IVM induces the imbalance in Cl
-ion
--- 49
Figure 23. Cl
-channel blockers inhibit IVM -induced ER
vacuolation and cell death
--- 50
Figure 24. Cl
-channel blocker inhibits IVM -induced ER
vacuolation and permeabilization
--- 51
Figure 25. Inhibition of Cl
-channels may act downstream of
IVM-induced ER stress and Ca
2+imbalance
--- 52
Figure 26. IVM induces mitochondria dilation
--- 61
Figure 27. IVM induces the loss of mitochondrial membrane
potential
--- 62
Figure 28. induced ROS generation is not important for
IVM-induced ER vacuolation and cell death
--- 63
Figure 29. IVM decreases the basal respiration and ATP synthesis
--- 64
Figure 30. Ivermectin induces the release of Cl
-enriched in
mitochondria
--- 65
Figure 31. DIDS and NA inhibit the imbalance of Cl
-and loss of
- 1 -
I. INTRODUCTION
[Ivermectin (IVM)]
Ivermectin, a macrocyclic lactone derived from Streptomyces avermitilis, is FDA-approved antiparasitic agent (Crump A, 2017; Bai SH et al., 2016). It has been utilized by millions of people around the world exhibiting a wide margin of clinical safety. In parasites and helminths, ivermectin increases the activity of γ-aminobutyric acid (GABA) receptors or glutamate-gated chloride ion channels (Glu-Cl), which blocks the signal between neuron and muscle (Bai SH et al., 2016). In mammals, GABA-sensitive neurons are secured by the blood-brain barrier (BBB) within the central nervous system (CNS), protecting vertebrates against potentially harmful effects of IVM (Bai SH et
al., 2016; Juarez M et al., 2018). IVM was reported that can inhibit the SERCA,
sarcoplasmic/endoplasmic-reticulum Ca2+-ATPase. Recently, ivermectin was reported to have an anticancer effect, inducing autophagy or apoptosis in various cancer cell lines, including breast cancer, glioma, colon cancer and leukemia (Markowska A et al., 2019; Juarez M et al., 2018; Dou Q et al., 2016; Liu Y et al., 2016; Melotti A et al., 2014; Sharmeen S et al., 2010; Draganov D et al., 2015). In addition, IVM induced mitochondrial dysfunction, which is caused by the generation of ROS or inhibition of respiratory complex I (Juarez M et al., 2018; Liu Y et al., 2016; Draganov D et al., 2015). Furthermore, IVM was induced immunogenic cell death by activation of P2X4/P2X7/Pannexin- 1 pathway (Draganov D et al., 2015), and it also inhibited the WNT-TCF pathway to suppress cancer proliferation (Melotti A et al., 2014).
- 2 -
[Breast cancer]
Breast cancer is the most common malignancy in women, and one of the three most common malignancies worldwide, along with lung and colon cancer (Ferlay J et al., 2015; Torre LA et al., 2012). In 2015 there were 2.4 million estimated new cases and 523,000 estimated deaths worldwide in women, which correspond to about 29% of the total incident cancer cases and 14% of all cancer deaths (Fitzmaurice C et al., 2015). Breast cancer cells may overexpress specific receptors (estrogen receptor (ER), progesterone receptor (PR), human epidermal growth factor receptor 2 (HER2)) which, when activated can initiate downstream signaling resulting in the expression of genes for cancer cell proliferation, growth, survival, migration, angiogenesis and other vital cell cycle pathways (Pal SK et al., 2011; Yarden Y et al., 2001). Triple-negative breast cancer (sometimes abbreviated TNBC) refers to any breast cancer that is defined by the lack of expression of ER, PR, and HER2 (Gluz O et al., 2009). Thus, TNBC is characterized by its unique molecular profile, aggressive nature, distinct metastatic patterns, and lack targeted therapies (Perou CM, 2011). Population-based studies have also demonstrated similar results with reduced breast cancer specific survival among those with TNBC, as compared with the luminal subtype (Carey LA et al., 2006). Although triple negative breast cancers are associated with a generally poor breast cancer specific outcome. The conventional route to treat TNBC patients by taxol derivatives and anthracycline chemotherapy is still widely used until more “druggable” targets are identified (Gluz O et al., 2009). TNBC are resistant to anthracycline and taxane drugs, thus, it is urgently needed to develop new drugs with better efficiency and fewer side effects.
- 3 -
[ER stress]
The endoplasmic reticulum (ER) is a major organelle for protein translocation, protein folding, and protein post-translational modifications that allow further transport of proteins to the Golgi apparatus and ultimately to vesicles for secretion or display on the plasma surface (Sano R et al., 2013). The folding capacity of the ER is limited by tightly regulated expression of protein chaperones such as glucose related protein 78 (GRP78), GRP94, and calreticulin (CRT). The ER lumen has an oxidative environment, which is crucial for the formation of disulfide bonds via protein disulfide isomerase (PDI). It is thus understandable that many physiological and/or environmental perturbations including alterations in Ca2+ homeostasis, redox changes (Delic
et al., 2012), accumulation of misfolded and aggregating proteins (Ellgaard
and Helenius, 2001), elevated secretory protein synthesis, glucose deprivation (Ikesugi et al., 2006; Auf et al., 2010; Badiola et al., 2011), altered N -glycosylation (Olivari and Molinari, 2007), cholesterol overload (Kedi et al., 2009; Li et al., 2009), ischaemia (Bilecova-Rabajdova et al., 2010) and viral infection (Zhang and Wang, 2012), might cause an ER stress (Vannucal K et
al., 2013). Physiological or pathological insults which overwhelm the folding
capacity of the ER, a process named “ER-stress”, activate an evolutionary conserved cascade of signaling events known as the ER stress response, or unfolded protein response (UPR), a tightly orchestrated collection of intracellular signal transduction reactions designed to restore protein homeostasis (Ojha R et al., 2017). The UPR is mediated by three molecular sensors present on the membrane of the endoplasmic reticulum; PKR-like ER kinase (PERK), activated transcription factor 6 (ATF6) and inositol-requiring
- 4 -
enzyme 1 alpha (IRE1) (Wang M et al., 2016). The ER resident BiP (Hsp70) binds the luminal tails of PERK, ATF6, and IRE1 to suppress their activity. When the levels of unfolded protein are elevated, BiP is titrated away, enhancing unfolded protein response (Bertolotti A et al., 2000; Shen J et al., 2002). BiP dissociates from UPR sensors, ATF6, IRE1, and PERK, in the presence of unfolded proteins. ATF6 translocates to the Golgi apparatus where it is subsequently cleaved by S1P and S2P. The cytosolic domain translocates to the nucleus where it promotes transcription of BiP, XBP1, proteins involved in lipid biosynthesis, and other chaperones. IRE1 and PERK both are oligomerized and auto-phosphorylated after dissociating from BiP. IRE1 splices incoming mRNAs, including Xbp1. Spliced Xbp1 is translated (XBP1s) and promotes transcription of BiP, EDEM1, proteins involved in lipid biosynthesis, and other chaperones. The product of unspliced Xbp1 translation (XBP1u) inhibits other components of the UPR, specifically XBP1s and ATF6. Phosphorylated PERK, in turn, phosphorylates eIF2α, inhibiting general translation. Nevertheless, some proteins, such as ATF4, are preferentially translated. ATF4 promotes transcription of CHOP, GADD34, and proteins involved with amino acid regulation, redox homeostasis, and apoptosis (Lewy TG et al., 2017).
- 5 -
[ER reorganization]
The ER forms a contiguous structure of interconnected sheets and tubules that spreads from the nuclear envelope to the cell cortex (Chen S et al., 2013). ER reorganization is a structure characterized by aggregated or stacked ER membrane (Erik L. Snapp et al., 2003). ER reorganization is a reversible structure (Erik L. Snapp et al., 2003), and it can be removed by autophagy (Li X et al., 2016). ER reorganization is one of the physiologically redundant response and is known to be associated with diseases such as Emery-Dreifuss case, torsion dystonia, and Hodgkin's lymphoma (Fidziańska A et al., 2004; Gonzalez-Alegre P et al., 2004; Parmley RT et al., 1976). ER reorganization is previously reported that it was induced by overexpression of ER membrane protein, including Cytochrome b(5)-GFP, Calnexin-GFP, STIM, ORAI3, and VAPB mutant, or by knockdown of Mcl-1, Syntaxin 18, a-SNAP (Erik L. Snapp et al., 2003, Li X et al., 2016, Varadarajan S et al., 2012; Varadarajan S et al., 2013; Korkhov VM et al., 2009). And some chemicals such as Apogossypol (pan-Bcl-2 antagonist), Calmidazolium and A-7 (calmodulin antagonist), Thapsigargin, Cyclopiazonic acid, and 2,5-di-t-butyl-1,4-benzohydroquinone (SERCA inhibitor), PDMP (glucosylceramide synthase and sphingomyelin synthase inhibitor) were also reported that induced ER reorganization (Erik L. Snapp et al., 2003; Varadarajan S et al., 2012). Imbalance of ion homeostasis (Ca2+, Na+) and disruption of vesicle trafficking
are also one of the causes of ER reorganization. (Varadarajan S et al., 2012; Varadarajan S et al., 2013; Korkhov VM et al., 2009). In addition, as a result of connectivity mapping analysis conducted using Apogossypol, it was confirmed that ER reorganization was also caused by various compounds such as ivermectin (anti-parasitic drug), thapsigargin (SERCA inhibitor),
- 6 -
nordihydroguaiaretic acid (SERCA inhibitor, antioxidant), astemizole (antihistamine drug), troglitazone (antidiabetic and anti-inflammatory drug), mefloquine (anti-malaria drug), suloctidil (withdrawn peripheral vasodilators), terfenadine (anti-histamine drug) (Varadarajan S et al., 2012). Furthermore, ER reorganization is known to attenuate the ER to Golgi membrane trafficking and induce ER stress and UPR (Varadarajan S et al., 2012; Varadarajan S et
al., 2013), but underlying mechanisms bout ER reorganization still remain
- 7 -
[ER vacuolation]
ER vacuolation is the structure that have a balloon-like shape due to ER dilation. The vacuolization of ER components seems to be due to the osmotic pressure increase resulting from high quantities of misfolded proteins in the ER (Andrey V et al., 2016). The compensation of osmotic pressure by water diffusion into the ER leads to the formation of vacuoles. Furthermore, dysfunction of big conductance calcium-activated potassium channels (BKCa) induced ER vacuolation (Hoa N et al., 2009). BKC inhibition was induced increase of K+ in ER and mitochondria, and the K+ mediated by osmotic effects leads to the formation of vacuoles (Bury M et al., 2013). Collectively, increase of osmotic pressure in ER via accumulation of misfolded protein or ion imbalance can lead to the ER vacuolation (Andrey V et al., 2016). And some chemicals such as curcumin, celastrol, ophiobolin A that can induce ER stress when it was treated with the cancer cells (Lee D et al., 2016). In Zika virus infection, heme depletion, Also, these are reported that ER vacuolation was appeared (Monel B et al., 2017; Petrillo S et al., 2018). In addition, dysfunctional ERAD, such as treatment of proteasome inhibitor, HSP90 inhibition, overexpression of dominant negative VCP, can affect the intracellular proteostasis and induced ER vacuolation (Mimnaugh EG, 2006). Moreover, ER vacuolation is phenotype of paraptosis that is non-apoptotic cell death mode accompanied the ER and mitochondria dilation (Lee D et al., 2016). Therefore, induction of ER vacuolation can lead the cytotoxicity.
- 8 -
[ER permeabilization]
ER permeabilization is a phenomenon that induced leakage of ER luminal protein from ER to cytosol caused by loss of ER membrane integrity. According to previous studies, ER membrane permeabilization depend on the proapoptotic Bcl-2 members Bax and Bak (Wang X et al., 2011). Under ER stress condition by palmitate, suppression of IRE1 signaling led to the accumulation of the BH3 domain-containing protein Bnip3, which in turn triggered the oligomerization of Bax and Bak in the ER membrane and ER membrane permeabilization (Kanekura K et al., 2015; Kanekura K et al., 2015). ER permeabilization can be assessed using stable cell line expressed ER luminal fluorescent protein S the ER-localized fluorescent protein is released into the cytosol and occupies all the cellular space in cells undergoing ER stress (Wang X et al., 2011; Kanekura K et al., 2015; Kanekura K et al., 2015). Homeostatic alterations in the ER play roles in the pathogenesis of chronic human disorders, such as type 1 and type 2 diabetes, myocardial infarction, stroke, and neurodegeneration, as well as inherited disorders including Wolfram syndrome, which is characterized by b cell death and neurodegeneration (Wang S et al., 2012; Fonseca SG et al., 2011). The ER membrane permeabilization occur in acquired pathological states associated with ER dysfunction, namely, ischemia/reperfusion injury and stroke, as well as Wolfram syndrome, an inherited disease state (Kanekura K et al., 2015)
- 9 -
[Paraptosis]
The most current anticancer therapies, including chemo-, radio‐ , and immuno-therapy, primarily activate the apoptosis in cancer cells (Fulda S, 2009). But inherent or acquired resistance of cancer cells to various proapoptotic treatments leads to therapeutic failure (Moulder S, 2010). Thus, a better understanding of alternative, non-apoptotic cell death pathways may facilitate the design of novel therapeutics against malignant cancer cells. Paraptosis is one of programmed cell death mode accompanied by dilation of the ER and mitochondria. It lacks the apoptotic features, including activation of caspase, DNA fragmentation, chromatin condensation, and the formation of apoptotic body. Several reports have shown that paraptosis requires de novo protein synthesis, activation of mitogen-activated protein kinases (MAPKs), and AIP-1/Alix decrease (Sperandio S et al., 2004). Also, recent reports demonstrated that ionic imbalance of Ca2+ (Yoon MJ et al., 2012; Yoon MJ et
al., 2014) and K+ (Bury M et al., 2013), generation of ROS (Yoon MJ et al., 2010; Ghosh K et al., 2016), and perturbation of cellular proteostasis due to proteasomal inhibition (Yoon MJ et al., 2010; Yoon MJ et al., 2014), and disruption of sulfhydryl homeostasis (Kar R et al., 2009; Kim IY et al., 2017; Seo MJ et al., 2019) are involved in the paraptosis. However, the underlying mechanism of paraptosis for triggering dilation of ER and mitochondria is not fully understood.
- 10 -
Here, we show that IVM kills various cancer cells via paraptosis. Interestingly, the ER is structurally altered, including the ER reorganization, vacuolation, and permeabilization. IVM increases lipid droplet, relevant to IVM-induced ER reorganization, but not ER vacuolation, ER permeabilization, and cell death. IVM upregulates ER stress, and reduction of ER stress by CHX pretreatment effectively inhibits IVM-induced ER vacuolation, ER permeabilization, and cell death. In addition, IVM increases the intracellular Ca2+ and Cl-, and these related to IVM-induced ER vacuolation, ER
permeabilization, and cell death. Collectively, our results show that IVM treatment may provide a therapeutic strategy against breast cancer cell via induction of paraptosis mediated by ER stress and imbalance of Ca2+ and Cl
- 11 -
II. MATERIALS AND METHODS
A. Chemicals and antibodies
Chemicals and reagents were obtained as follows: IVM, 3-methyladenine (3-MA), bafilomycin A1 (Bafilo), chloroquine (CQ), necrostatin-1, 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA), 2,3-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis (acetoxymethyl ester) (BAPTA-AM), ethylenebis (oxyehylenenitrilo) tetraacetic acid (EGTA), 4,4′-Diisothiocyanatostilbene-2,2′-disulfonic acid disodium salt hydrate (DIDS), Niflumic acid, ruthenium red, and cycloheximide (CHX), oleic acid, Triacsin C from Sigma-Aldrich (St. Louis, MO, USA); SP00000, PD98059, U0126 and SB203580 from Calbiochem (EDM Millipore Corp., Billerica, MA, USA); Fluo-3-AM, and 4′,6-diamidino–phenylindole (DAPI) H2DCF-DA, tetramethylrhodamine
methyl ester (TMRM) from Molecular Probes (Eugene, OR, USA); z-VAD-fmk from R&D Systems (Minneapolis, MN, USA); ER-Tracker Blue-White DPX Invitrogen (Carlsbad, CA, USA). The following primary antibodies were used: β-actin (sc-47778), ATF4 (sc-200), Nogo (sc-271878) from Santa Cruz Biotechnology (Santa Cruz, CA, USA); CHOP/GADD153 (#2895), PERK (#5683), and IRE1 (#3294), phospho-eIF2α (#9721), and eIF2α (#9722) from Cell Signaling Technology (Danvers, MA, USA); 113) from Enzo Life Sciences (Farmingdale, NY, USA);BAP31 (ab37120) from Abcam (Cambridge, UK). The secondary antibodies, including rabbit IgG HRP (G-21234) and mouse IgG HRP (G-21040), were obtained from Molecular Probes.
- 12 -
B. Cell culture
MDA-MB-435S, MDA-MB 468 (breast cancer), MCF-10A (non-tumorigenic breast epithelial cell line), Panc-1, BxPC-3 (pancreatic cancer), A549, NCI-H460 (lung cancer), Huh-7 (hepatocellular carcinoma) from the American Type Culture Collection (ATCC, Manassas, VA, USA). SNU-449 (hepatocellular carcinoma) cells were obtained from the Korean Cell Line Bank (KCLB, Seoul, Korea). MDA-MB 435S, Panc-1, Huh-7 cells were cultured in DMEM supplemented with 10% fetal bovine serum (FBS) and 1% antibiotics (GIBCO-BRL, Grand Island, NY, USA). MDA-MB 468, BxPC-3, A549, NCI-H460, SNU-449 cells were cultured in RPMI-1640 medium supplemented with 10% FBS and 1% antibiotics. MCF-10A cells were maintained in DMEM/F12 medium supplemented with 10% FBS and 1% antibiotics. Cells were incubated in 5% CO2 at 37 °C.
C. Measurement of cellular viability
Cell viability was assessed by double labeling of cells with 2 μM calcein-AM and 4 μM EthD-1. The calcein-positive live cells and EthD-1-positive dead cells were visualized using an IncuCyte device (Essen Bioscience, Ann Arbor, MI, USA) and analyzed using the IncuCyte ZOOM 2016B software. The percentage of live cells was normalized to that found in untreated control cultures (100%).
- 13 -
D. Immunoblot analyses
Cells were washed in PBS and lysed in boiling sodium dodecyl sulfate– polyacrylamide gel electrophoresis (SDS–PAGE) sample buffer (62.5 mM Tris (pH 6.8), 1% SDS, 10% glycerol, and 5% β-mercaptoethanol). The lysates were boiled for 5 min, separated by SDS–PAGE, and transferred to an Immobilon membrane (Millipore). After nonspecific binding sites were blocked for 1 h using 5% skim milk, the membranes were incubated for 2 h with specific antibodies. Membranes were then washed three times with Tris-Buffered Saline Tween-20 (TBST) and incubated further for 1 h with horseradish peroxidase-conjugated anti-rabbit, -mouse, or -goat antibody. Visualization of protein bands was accomplished using ECL (Amersham Life Science). The fold change of each target protein level compared to β-actin was determined by densitometric analysis.
E. Morphological examination of the ER and mitochondria
Examination of the stable cell lines expressing the fluorescence specifically in mitochondria or the endoplasmic reticulum. To establish the stable cell lines expressing the fluorescence specifically in mitochondria or the ER, MDA-MB 435S cells were transfected with the pEYFP-Mito or pEYFP-ER vector (Clontech, Mountain View, CA, USA) and Sec61β-GFP (Addgene, ,). Stable cell lines expressing pEYFP-Mito or pEYFP-ER or Sec61β -GFP (YFP-Mito or YFP-ER) were selected with fresh medium containing 500 μg/ml G418 (Calbiochem). After treatments, the morphological changes of the ER and mitochondria were observed under fluorescence microscopy (Carl Zeiss) or a K1-Fluo confocal laser-scanning microscope (Nanoscope Systems. Daejeon, Korea).
- 14 -
F. Immunofluorescence microscopy
After treatments, the cells were fixed with acetone/methanol (1:1) for 5 min at −20 °C and blocked in 5% BSA in PBS for 30 min. Fixed cells were incubated overnight at 4 °C with primary antibodies diluted in PBS [anti-BAP31(1:500, rabbit, Abcam), anti-RTN4 (1:500, mouse, novos)], washed three times in PBS, and incubated for 1 h at room temperature with anti-mouse or anti-rabbit Alexa Fluor 488 or 594 (1:1000, Molecular Probes). Slides were mounted with ProLong Gold antifade mounting reagent (Molecular Probes) and cell staining was visualized with the K1-Fluo confocal laser scanning microscope.
G. Transmission electron microscopy
Cells were pre-fixed in Karnovsky’s solution (1% paraformaldehyde, 2% glutaraldehyde, 2 mM calcium chloride, 0.1 M cacodylate buffer, pH 7.4) for 2 h and washed with cacodylate buffer. Post-fixing was carried out in 1% osmium tetroxide and 1.5% potassium ferrocyanide for 1 h. After dehydration with 50–100% alcohol, the cells were embedded in Poly/Bed 812 resin (Pelco, Redding, CA, USA), polymerized, and observed under an electron microscope (EM 902A, Carl Zeiss, Oberkochen, Germany).
H. Measurement of intracellular Ca2+ levels
To measure cytosolic Ca2+ levels ([Ca2+]c), treated cells were incubated with 1 μM Fluo-3-AM at 37 °C for 20 min, washed with HBSS (without Ca2+
or Mg2+), and visualized with the K1-Fluo confocal laser scanning microscope.
- 15 -
I. Measurement of intracellular Cl- levels
To measure intracellular Cl- level, treated cells were incubated with 300 μM MQAE at 37 °C for 20 min, washed with HBSS (without Ca2+ or Mg2+),
and visualized with Axiovert 200M fluorescence microscopy (Carl Zeiss).
J. Measurement of intracellular reactive oxygen species levels
To measure intracellular ROS level, treated cells were incubated with 300 μM MQAE at 37 °C for 20 min, washed with HBSS (without Ca2+ or Mg2+),
and visualized with Axiovert 200M fluorescence microscopy (Carl Zeiss).
K. Measurement of Cellular Oxygen Consumption Rate
To determine cellular dependency on glycolysis or mitochondrial respiration and cellular endogenous oxygen consumption rate (OCR) were measured using Seahorse XF24 analyzer (Seahorse Bioscience Inc.) according to the manufacturer's protocol. Briefly, cells were plated at a density of 10,000 cells/well on XF24 tissue culture plate and preincubated with XF assay medium (Seahorse Bioscience) containing 1 mM pyruvate and 5 mM glucose in the presence or absence of 2 mM oligomycin (Oli) or 25 mM 2-deoxyglucose (DOG) for 1 h, and further equilibrated with the pre-calibrated probe inside the instrument for 2 h. Then, endogenous OCR was measured under basal condition and normalized to cell number. OCR was expressed as percent of the values obtained from untreated control cells.
- 16 -
L. ATP measurement
Steady‐state levels of ATP were estimated using the Cell Titer‐Glo from Promega Corporation (Madison, Wisconsin, USA), according to manufacturer's instructions. In brief, 1 × 104 cells per well of MDA-MB 435S cells were plated on 96‐well plates. After 24 h, cells were treated with 25 μM IVM. After treatment of IVM for 24 h, the plates containing the cells were equilibrated to room temperature for 30 min prior to addition of the luciferin/luciferase/cell lysis mixture. Absolute luminescence from quadruplicate experiments was recorded using a Synergy H1 Hybrid Multi-Mode Microplate Reader from Biotek (Winooski, VT, USA).
M. Statistical analysis
All data were presented as mean ± S.D. (standard deviation) from at least three separate experiments. Student’s t test was applied to evaluate the differences between treated and control groups with cell viability. Data from multiple groups were analyzed by one-way ANOVA, followed by Bonferroni multiple comparison test. For all the tests, the level of significance was values of P < 0.05.
- 17 -
III. RESULTS
1. Ivermectin (IVM) effectively induces cell death in various cancer cells
To investigate the anti-cancer effect of IVM in cancer cells, we treated with IVM in breast cancer cells (MDA-MB 435S and MDA-MB 468) and human breast non-tumorigenic epithelial cell line (MCF-10A) as a counterpart. The cytotoxicity of IVM was assessed using Live and Dead assay kit, which demonstrated that IVM dose-dependently decreased cellular viability in MDA-MB 435S and MDA-MB 468 cells, but not MCF-10A cells (Figure 1A). Interestingly, the cell death induced by IVM was commonly accompanied by cytoplasmic vacuolation in MDA-MB 435S 25 μM and MDA-MB 468 20 μM, but not in a normal cell at doses up to 50 μM of IVM (Figure 1B). Next, we further tested the effects of IVM on other types of cancer cells, which isolated in pancreas, liver, and lung, respectively; pancreatic cancer cells (BxPC-3 and Panc-1), hepatocellular carcinoma (Huh-7 and SNU449), and lung cancer (A549 and H460) (Figure 2A). These results reveal that IVM effectively induced cell death in all the tested cancer cells. In addition, when IVM treatment of each cell line, cytoplasmic vacuolation was occupied all the cellular space except nucleus (Figure 2B). To identify the cell death mode induced by IVM, we tested the effects of diverse inhibitors including z-VAD-fmk (z-VAD; apoptosis inhibitor), Necrostatin-1 (Nec-1; necroptosis inhibitor), and 3-methyladenine, bafilomycin A1, chloroquine (3-MA, Bafilo, CQ; autophagy inhibitor). But all these inhibitors did not inhibit the cytoplasmic vacuolation as well as the cell death induced by IVM (Figure 3 and 4). Taken together, our results reveal that IVM induced cancer cell death accompanied
- 18 -
by cytoplasmic vacuolation, and neither apoptosis, necroptosis, nor autophagic cell death was involved in the cell death induced by IVM.
- 19 -
Figure 1. IVM induces the cell death accompanied by cytosolic vacuolation in breast cancer cells, but not in normal breast cells. (A) Cells
were treated with the indicated concentrations of ivermectin for 24 h and cellular viability was assessed using calcein-AM and EthD-1. Data represent the mean ± SD. One-way ANOVA and Bonferroni’s post hoc test. *P < 0.005 vs. untreated control. (B) Cells were treated with the indicated concentrations of ivermectin for 12 h and observed under phase-contrast microscope. Scale Bars, 20 μM.
- 20 -
Figure 2. IVM induces cytoplasmic vacuolation and the cell death in various cancer cells. (A) Cells were treated with the indicated concentrations
of ivermectin for 24 h and cellular viability was assessed using calcein-AM and EthD-1. Data represent the mean ± SD. One-way ANOVA and Bonferroni’s post hoc test. *P < 0.005 vs. untreated control. (B) Cells were treated with each IC50 concentrations of ivermectin for 12 h and observed
- 21 -
Figure 3. Neither apoptosis nor necroptosis is involved in IVM-induced cell death. (A, B) MDA-MB 435S Cells were pretreated with the indicated
concentrations of z-VAD-fmk (an apoptosis inhibitor) or Necrostatin-1 (a necroptosis inhibitor) for 30 min and further treated with 25 μM IVM for 24 h. (A) Cellular viability was assessed using calcein-AM and EthD-1. Data represent the mean ± SD. One-way ANOVA and Bonferroni’s post hoc test. *P < 0.005 vs. untreated cells. (B) Cells were observed by phase-contrast microscopy. Scale Bars, 20 μm.
- 22 -
Figure 4. Autophagy is not involved in IVM-induced cell death. (A)
MDA-MB 435S cells were pretreated with the indicated concentrations of autophagy inhibitors (3-MA, Bafilomycin A, or chloroquine) for 30 min and further treated with 25 μM IVM for 24 h, and cellular viability was assessed using calcein-AM and EthD-1. Data represent the mean ± SD. One-way ANOVA and Bonferroni’s post hoc test. *P < 0.005 vs. untreated cells, #P < 0.005 vs. cells treated with IVM. (B) MDA-MB 435S Cells were pretreated with autophagy inhibitors (3-MA, Bafilomycin A, or chloroquine) for 30 min, treated with 25 μM IVM for 24 h, and observed by phase-contrast microscopy. Scale Bars, 20 μm.
- 23 -
2. IVM induces catastrophic changes in the endoplasmic reticulum structure
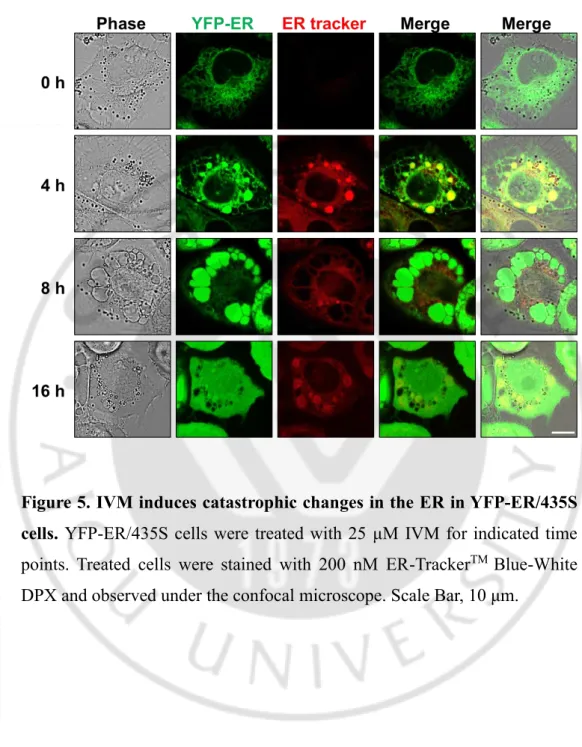
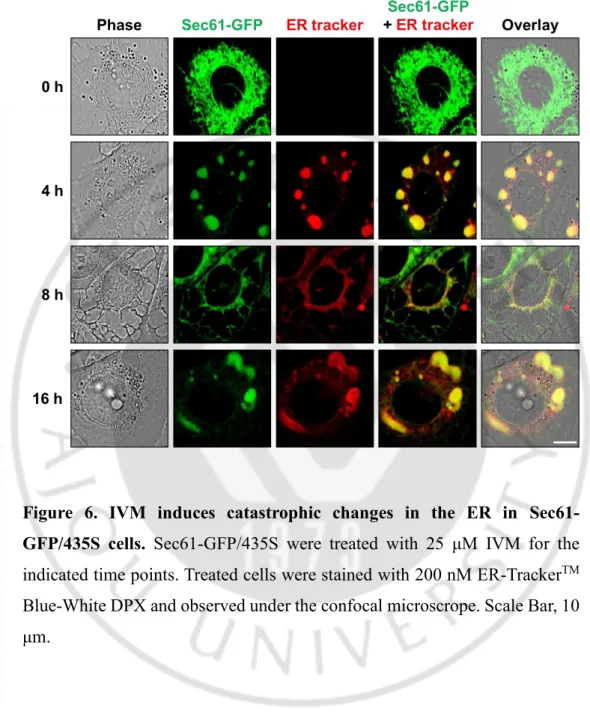
Next, to further evaluate the cell death mode induced by IVM, we examined the origins of the IVM-induced vacuoles. Since autophagy inhibitors did not alter IVM-induced vacuolation, and the morphological features of these vacuoles were similar to dilation of the ER, we hypothesize the origins of vacuoles might be the ER. To confirm our speculation, we employed MDA-MB 435S sublines stably expressing the pEYFP-ER (YFP-ER) and pSec61β-GFP (Sec61-pSec61β-GFP) which represent ER lumen and membrane, respectively. When we observed YFP-ER cells and Sec61-GFP cells stained with ER-Tracker Blue-White DPX under the confocal microscope, both YFP-ER and Sec61-GFP showed reticular ER structure in untreated cells with weak red fluorescence intensity. Interestingly, even though cytoplasmic vacuolation was not observed in IVM treated cells for 4 h in phase contrast image, YFP-ER/ER-Tracker White DPX and Sec61-GFP/ER-Tracker Blue-White DPX was co-localized in aggregated structure, suggesting that the ER was stacked and it means that ER reorganization was induced by IVM (Varadarajan S et al., 2012). At 8 h, when the cytoplasmic vacuolation was observed in the phase contrast image, simultaneously only YFP-ER occupied within numerous cytoplasmic vacuolation, and the fluorescence of Sec61-GFP and ER-tracker were localized in the surface of these vacuoles. In cells treated with IVM for 16 h, cytoplasmic vacuolation disappeared in the phase contrast image, YFP-ER fluorescence was diffused in all the cellular space including the nucleus, and Sec61-GFP and ER-tracker have shown the re-formed ER aggregation structure (Figure 5, 6). These results suggest that the membrane integrity of the ER was lost, and it’s permeabilization induced leakage of the
- 24 -
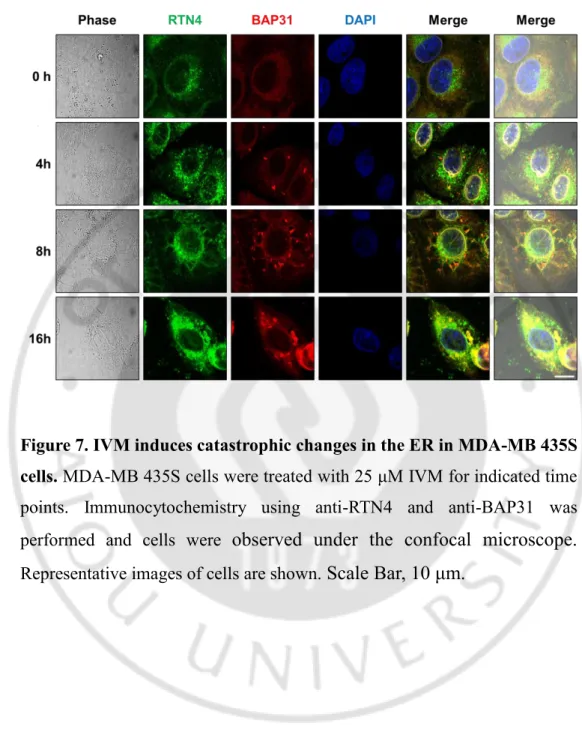
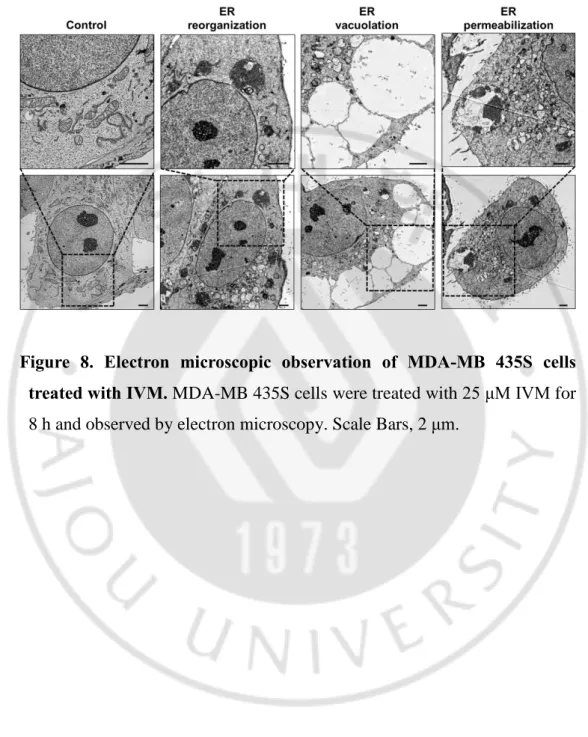
luminal proteins (Kanekura K et al., 2015). These results were further confirmed using immunocytochemistry of BAP31and RTN4, the ER membrane proteins (Figure 7). In addition, electron microscopy (EM) further revealed that IVM induced aggregated ER; ER reorganization, and dilated ER; ER vacuolation in IVM treated cells (Figure 8). Furthermore, the time-lapse images of IVM-treated YFP-ER cells using IncuCyte have shown that the ER, in turn, was reorganized, vacuolated, and permeabilized by IVM treated cells (Figure 9A). The quantification of the ER morphologies demonstrated that occurred in turn prior to cell death (Figure 9B). Collectively, out results indicated that IVM induces cell death accompanied by catastrophic changes of the ER structure; in the order of ER reorganization, vacuolation, and permeabilization.
- 25 -
Figure 5. IVM induces catastrophic changes in the ER in YFP-ER/435S cells. YFP-ER/435S cells were treated with 25 μM IVM for indicated time
points. Treated cells were stained with 200 nM ER-TrackerTM Blue-White
- 26 -
Figure 6. IVM induces catastrophic changes in the ER in Sec61- GFP/435S cells. Sec61-GFP/435S were treated with 25 μM IVM for the
indicated time points. Treated cells were stained with 200 nM ER-TrackerTM
Blue-White DPX and observed under the confocal microscrope. Scale Bar, 10 μm.
- 27 -
Figure 7. IVM induces catastrophic changes in the ER in MDA-MB 435S cells. MDA-MB 435S cells were treated with 25 μM IVM for indicated time
points. Immunocytochemistry using anti-RTN4 and anti-BAP31 was performed and cells were observed under the confocal microscope.
- 28 -
Figure 8. Electron microscopic observation of MDA-MB 435S cells treated with IVM. MDA-MB 435S cells were treated with 25 μM IVM for
- 29 -
Figure 9. Time-lapse imaging of YFP-ER cells treated with induced by IVM. (A) YFP-ER/435S cells were treated with 25 μM IVM for the indicated
time points and observed by IncuCyte. Scale Bar, 20 μm. (b) YFP-ER/435S cells were treated with 25 μM IVM for the indicated time points. The percentages of cells with the respective ER structure were quantitated.
- 30 -
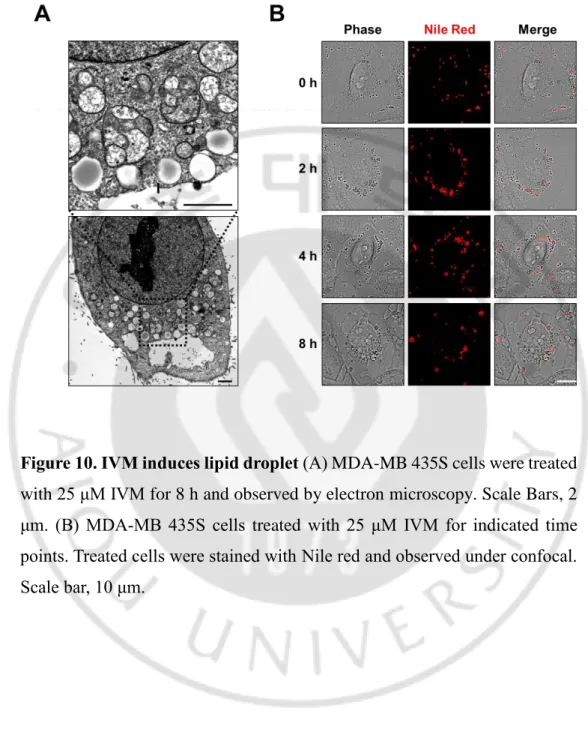
3. ER reorganization is associated with lipid
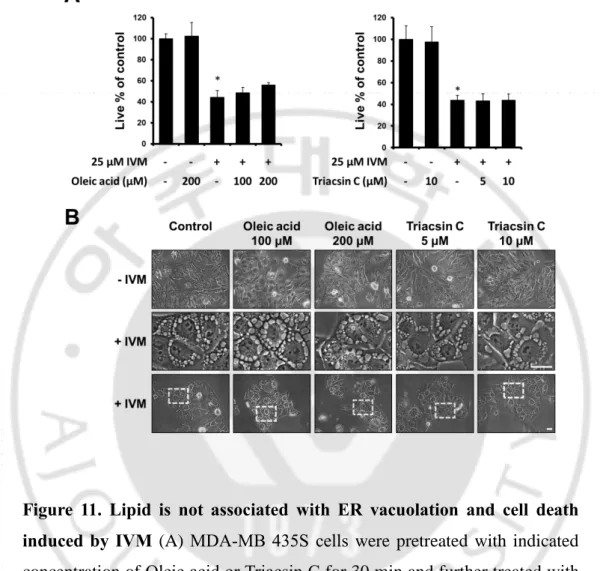
From electron microscopy, we observed lipid droplet-like vesicle (Figure 10A). Lipid droplets are lipid-rich cellular organelles that regulate the storage and hydrolysis of neutral lipids and formed at the endoplasmic reticulum (ER) membrane. (Mishra S et al., 2016). Thus, we speculated whether IVM-induced changes in the ER structure might be related to lipid droplet. To confirm the IVM-induced formation of lipid droplet, we stained IVM-treated MDA-MB 435S cells with Nile Red. We found that the numbers and sizes of lipid droplets were transiently increased with peak at 2 h of IVM treatment (Figure 10B). Thus, we tested the effect of regulators of lipid droplet on the IVM-induced ER structure changes. For this purpose, we used oleic acid, a fatty acid which stimulates lipid droplet formation (Lucía C. Lagrutta et al., 2017), and triacsin C, which reduces lipid droplet formation (Dechandt CRP et al., 2017). Lipid droplet was increased in cells pretreated with oleic acid and decreased in cells pretreated with Triacsin C, but both have not affected IVM induced cytoplasmic vacuolation and cell death (Figure 11A, B). Furthermore, to confirm whether lipid droplet affects IVM-induced changes of the ER structure, we pretreated with oleic acid or Triacsin C and further treated with IVM in YFP-ER cells. Interestingly, pretreatment with oleic acid increased the ER reorganization, and Triacsin C decreased the ER reorganization, but both did not alter the ER vacuolation and permeabilization (Figure 12). Taken together, these results suggested that ER reorganization was associated with lipid homeostasis, but it was irrelevant to ER vacuolation, permeabilization, and subsequent cell death.
- 31 -
Figure 10. IVM induces lipid droplet (A) MDA-MB 435S cells were treated
with 25 μM IVM for 8 h and observed by electron microscopy. Scale Bars, 2 μm. (B) MDA-MB 435S cells treated with 25 μM IVM for indicated time points. Treated cells were stained with Nile red and observed under confocal. Scale bar, 10 μm.
- 32 -
Figure 11. Lipid is not associated with ER vacuolation and cell death induced by IVM (A) MDA-MB 435S cells were pretreated with indicated
concentration of Oleic acid or Triacsin C for 30 min and further treated with 25 μM IVM for 24 h, and cellular viability was assessed using calcein-AM and EthD-1. Data represent the mean ± SD. One-way ANOVA and Bonferroni’s post hoc test. *P < 0.005 vs. untreated cells. (B) MDA-MB 435S cells were pretreated with indicated concentration of Oleic acid or Triacsin C for 30 min, and further treated with 25 μM IVM for 12 h, and observed by phase-contrast microscopy. Scale bar, 20 μm.
- 33 -
Figure 12. ER reorganization is associated the lipid. YFP-ER/435S cells
were pretreated with indicated concentration of Oleic acid or Triacsin C for 30 min, and further treated with 25 μM IVM for indicated time points and observed under the fluorescent and phase contrast microscope. Scale bar, 20 μm.
- 34 -
4. IVM induces paraptosis in MDA-MB 435S
Since the ER-derived vacuolation is a key morphological feature of paraptosis, we next tested whether IVM induces biochemical features of paraptosis. Since the ER vacuolation during paraptosis was shown to be associated with ER stress, we first examined the expression of ER stress-related proteins following treatment of MDA-MB 435S cells with IVM. We found that IVM treatment time-dependently upregulated the ER stress marker proteins, including IRE1α, ATF4, CHOP, phosphorylated PERK, and phosphorylated eIF2α in MDA-MB 435S cells (Figure 13A). Since paraptosis is known to require protein synthesis, we first tested the effect of tested the effect of the protein synthesis inhibitor, cycloheximide (CHX). We found that CHX treatment markedly blocked IVM-induced upregulation of ER stress marker proteins (Figure 13B). In addition, CHX pretreatment effectively inhibited IVM-induced cell death (Figure 14A) as well as cytoplasmic vacuolation (Figure 14B). Furthermore, CHX effectively inhibited IVM-induced vacuolation and permeabilization of the ER, but not ER reorganization (Figure 15). Since MAP kinases were shown to be positively associated with paraptosis, we examined the changes in the activities of MAP kinases following IVM treatment. Western blot analysis showed the biphasic activation of ERKs and JNKs, but monophasic activation of p38 with a peak at 4 h of IVM treatment (Figure 16A). In addition, when we investigated the functional significance of various MAP kinases in IVM-induced cell death using the respective inhibitors, we found that inhibition of the ERK pathway (using PD98059 or U0126) and p38 pathway (using SB203580) significantly attenuated IVM-induced cell death (Figure 16B) and vacuolation in MDA-MB 435S cells (Figure 16C). In addition, pretreatment with these inhibitors in
- 35 -
YFP-ER cells delayed IVM-induced ER vacuolation and permeabilization (Figure 17). Collectively, these results suggest that IVM may induce paraptosis-associated cell death and ER stress and MAPK pathway may be critically involved in this process.
- 36 -
Figure 13. IVM induces ER stress, which is blocked by CHX. (A) Cell
extracts were prepared from MDA-MB 435S cells treated with 25 μM IVM for the indicated time points and western blotting of the proteins associated with ER stress was performed. β-actin was used as a loading control in western blots. (B) MDA-MB 435S cells pretreated with 2μM CHX for 30 min were further treated with 25 μM IVM for the indicated time points. Cell extracts were subjected to Western blotting of the indicated proteins, with β-actin used as a loading control.
- 37 -
Figure 14. CHX blocks IVM-induced ER vacuolation and cell death. (A)
MDA-MB 435S cells were pretreated with indicated concentration of CHX C for 30 min and further treated with 25 μM IVM for 24 h, and cellular viability was assessed using calcein-AM and EthD-1. Data represent the mean ± SD. One-way ANOVA and Bonferroni’s post hoc test. *P < 0.005 vs. untreated cells; #P < 0.005 vs. cells treated with IVM. (B) MDA-MB 435S cells were pretreated with the indicated concentrations of CHX for 30 min, and further treated with 25 μM IVM for 12 h, and observed by phase-contrast microscopy. Scale Bar, 20 μm.
- 38 -
Figure 15. CHX blocks ER vacuolation and permeabilization but not ER reorganization. (A) YFP-ER/435S cells were pretreated with 2 μM CHX for
30 min, and further treated with 25 μM IVM for indicated time points and observed under the fluorescent and phase contrast microscope. Scale Bar, 20 μm. (B) YFP-ER/435S cells were pretreated with 2 μM CHX for 30 min and further treated with 25 μM IVM for indicated time points. The percentages of the cells with the respective ER structure were quantitated.
- 39 -
Figure 16. Activation of ERK and p38 significantly contributes to IVM-induced vacuolation and cell death. (A) Cells extracts were prepared from
MDA-MB 435S cells treated with 25 μM IVM for the indicated time points and western blotting of three MAP kinases was performed. β-actin was used as a loading control in western blots. (B) MDA-MB 435S cells were pretreated with the indicated concentrations of the respective MAPK inhibitors for 30 min and further treated with 25 μM IVM for 24 h, and cellular viability was assessed using calcein-AM and EthD-1. Data represent the mean ± SD. One-way ANOVA and Bonferroni’s post hoc test. *P < 0.005 vs. untreated cells; #P < 0.005 vs. cells treated with IVM. (C) MDA-MB 435S cells were pretreated with the indicated concentration of MAPK inhibitors for 30 min, and further treated with 25 μM IVM for 12 h, and observed by phase-contrast microscopy. Scale Bars, 20 μm.
- 40 -
Figure 17. Inhibition of ERK and p38 delays IVM-induced ER vacuolation and permeabilization. (A) YFP-ER/435S cells were pretreated
with 20 μM U0126 or 20 μM SB for 30 min, and further treated with 25 μM IVM for indicated time points and observed under the confocal. Scale Bars, 10 μm. (B) YFP-ER/435S cells were pretreated with 20 μM U0126 or 20 μM SB for 30 min and further treated with 25 μM IVM for indicated time points. The percentages of the cells with the respective ER structure were quantitated.
- 41 -
5. Increase in intracellular Ca2+ critically contributes to IVM-induced
paraptosis
The endoplasmic reticulum (ER) is a large, dynamic structure that serves many roles in the cell, including protein synthesis, lipid metabolism, and Ca2+
storage. (Sano R et al., 2013). Previously, we found that disruption of intracellular Ca2+ homeostasis plays a critical role in paraptosis (Yoon MJ et
al., 2012; Yoon MJ et al., 2014). And it was reported that IVM can inhibit the
SERCA, sarcoplasmic/endoplasmic-reticulum Ca2+-ATPase Ca2+ pumps. So,
we first tested whether disruption of Ca2+ homeostasis is associated with
IVM-induced structural changes in the ER and subsequent cell death. When we first measured the intracellular Ca2+levels using Fluo-3, Ca2+ indicator, we found
that IVM markedly increased intracellular Ca2+ levels from 8 h and further
increased at 16 h of IVM treatment (Figure 18A). To investigate the functional significance of this increased Ca2+ in IVM-induced cellular responses, we
examined the effect of Ca2+ scavengers, including BAPTA and EGTA,
extracellular Ca2+ scavenger, and BAPTA-AM, intracellular Ca2+ scavenger.
Among the tested Ca2+ scavengers, only BAPTA-AM effectively blocked
IVM-induced increase in intracellular Ca2+ levels (Figure 18B). In addition,
only BAPTA-AM significantly inhibited IVM-induced cell death (Figure 19A) as well as ER vacuolation (Figure 19B). When we further examined the effect of BAPTA-AM on IVM-induces structural changes in the ER, we found that BAPTA-AM effectively attenuated IVM-induced ER vacuolation and permeabilization, but not ER reorganization, in YFP-ER (Figure 20). When we tested the correlation between ER stress and Ca2+ imbalance, interestingly,
BAPTA-AM slightly delayed IVM-induced CHOP upregulation without any marked effect on the levels of other ER stress markers (Figure 21). In contrast,
- 42 -
CHX effectively inhibited IVM-induced increase in Ca2+ levels, suggesting that Ca2+ imbalance may act downstream of ER stress in IVM-induced signaling pathways and cell death.
- 43 -
Figure 18. IVM increases intracellular Ca2+, which is inhibited by
BAPTA-AM. (A) MDA-MB 435S cells were treated with 25 μM IVM for
indicated time points. Treated cells were stained with 2.5 μM Fluo-3 and observed under Confocal, 10 μm. (B) MDA-MB 435S cells were pretreated with BAPTA-AM and/or treated with 25 μM IVM for indicated time points. Treated cells were stained with 2.5 μM Fluo-3 and observed under Confocal, 10 μm.
- 44 -
Figure 19. BAPTA-AM blocks IVM-induced ER vacuolation and cell death. (A) MDA-MB 435S cells were pretreated with the indicated
concentrations of Ca2+ chelators for 30 min and further treated with 25 μM
IVM for 24 h, and cellular viability was assessed using calcein-AM and EthD-1. Data represent the mean ± SD. One-way ANOVA and Bonferroni’s post hoc test. *P < 0.005 vs. untreated cells; #P < 0.005 vs. cells treated with IVM. (B) MDA-MB 435S cells were pretreated with the indicated concentrations of Ca2+ chelators for 30 min, and further treated with 25 μM IVM for 24 h, and observed by phase-contrast microscopy. Scale Bars, 20 μm.
- 45 -
Figure 20. BAPTA-AM blocks IVM-induced ER vacuolation and permeabilization. (A) MDA-MB 435S cells were pretreated with 10 μM
BAPTA-AM for 30 min and further treated with 25 μM IVM for the indicated time points. Treated cells were stained with 250 nM Fluo-3 and observed by confocal microscopy. Scale bars, 10 μm. (B) YFP-ER/435S cells were pretreated with 10 μM BAPTA-AM for 30 min and further treated with 25 μM IVM for indicated time points. The percentages of the cells with the respective ER structure were quantitated.
- 46 -
Figure 21. IVM-induced Ca2+ increase is associated with ER stress. (A) MDA-MB 435S cells pretreated with 10 μM BAPTA-AM for 30 min and further treated with 25 μM IVM for indicated time points. Cell extracts were subjected to Western blotting of the indicated proteins, with β-actin used as a loading control. (B) MDA-MB 435S cells pretreated with 2 μM CHX for 30 min and further treated with 25 μM IVM for 12 h. The image was observed under the confocal microscopy. Scale Bars, 20 μm.
- 47 -
6. Imbalance in intracellular chloride may be involved in IVM-induced paraptosis
Previously, IVM was shown to target the Cl- channels, such as γ-aminobutyric
acid (GABA) receptors or glutamate-gated Cl- ion channels (Glu-Cl) (Bai SH
et al., 2016), which are expressed only in the nerve system. In addition, recent
studies reported that IVM induces Cl--dependent membrane hyperpolarization
and cell death in leukemia cells, maybe via unknown Cl--channel irrelevant to
GABA receptor or Glu-Cl (Bai SH et al., 2016). Furthermore, the increase of the intracellular Cl- by activation of chloride intracellular channel-1 (CLIC1)
was shown to play a critical role in the paraptosis induced by a purified resin glycoside fraction (Zhu D et al., 2019). Thus, we tested whether imbalance intracellular Cl- ion is also involved in IVM-induced paraptosis. First, we
measured the changes in Cl- levels using MQAE, an indicator of intracellular Cl-. We found that while Cl- was weakly detected only in organelles with similar morphology of mitochondria, a remarkable increase in Cl- was observed in the cytosol, suggesting the release of Cl- from these organelles. To test whether IVM-induced vacuolation and cell death is linked to the activity of Cl- channels, we examined the effects of various inhibitors of Cl -channels. We found that only DIDS (anion exchange inhibitor) and niflumic acid (Ca2+-activated Cl- channel blocker) were inhibited cell death (Figure 23A) and ER vacuolation (Figure 23B). In particular, pretreatment with DIDS or niflumic acid inhibited IVM-induced ER vacuolation and permeabilization, but nor ER reorganization (Figure 24). When we further investigate the correlation among Cl- imbalance and other signals associated with
IVM-induced cell death, we found that either DIDS or niflumic acid did not block IVM-induced ER stress (Figure 25A) and IVM-induced increase in Ca2+ levels
- 48 -
(Figure 25B). These results suggest that Cl- channel may be activated downstream of ER stress and Ca2+, contributing to ER vacuolation, permeabilization and subsequent cell death.
In summary, IVM induces cancer cell death via catastrophic changes in the structure and function of ER. In this process, ER stress and imbalance of Ca2+ and Cl- critically contribute to the anticancer effect of IVM.
- 49 -
Figure 22. IVM induces the imbalance in Cl- ion. (A) MDA-MB 435S cells were treated with 25 μM IVM for indicated time points. Treated cells were stained with 3 mM MQAE and observed under the fluorescent and phase contrast microscope. Scale bars, 20 μm.
- 50 -
Figure 23. Cl- channel blockers inhibit IVM-induced ER vacuolation and
cell death. (A) MDA-MB 435S cells were pretreated with the indicated
concentrations of Cl- channel inhibitors for 30 min and further treated with 25 μM IVM for 24 h, and cellular viability was assessed using calcein-AM and EthD-1. Data represent the mean ± SD. One-way ANOVA and Bonferroni’s post hoc test. *P < 0.005 vs. untreated cells; #P < 0.005 vs. cells treated with IVM. (B) MDA-MB 435S cells were pretreated with the indicated concentrations of Cl- channel inhibitors for 30 min, and further treated with 25 μM IVM for 12 h, and observed by phase-contrast microscopy. Scale Bars, 20 μm.
- 51 -
Figure 24. Cl- channel blocker inhibits IVM-induced ER vacuolation and
permeabilization. (A) MDA-MB 435S cells were pretreated with the 200 μM
DIDS or 200 μM niflumic acid (NA) for 30 min, and further treated with 25 μM IVM for 12 h, and observed by confocal microscopy. Scale Bars, 10 μm.
- 52 -
Figure 25. Inhibition of Cl- channels may act downstream of IVM-induced
ER stress and Ca2+ imbalance. (A) MDA-MB 435S cells pretreated with 200 μM DIDS, 200 μM Niflumic acid (NA) for 30 min and further treated with 25 μM IVM for indicated time points. Cell extracts were subjected to Western blotting of the indicated proteins, with β-actin used as a loading control. (B) MDA-MB 435S cells pretreated with 200 μM DIDS and NA for 30 min and further treated with 25 μM IVM for 12 h. The image was observed under the fluorescent and phase contrast microscope. Scale Bars, 20 μm.
- 53 -
Discussion
Previously, IVM was reported that have anticancer effect in various cancer cells (Markowska A et al., 2019; Juarez M et al., 2018; Dou Q et al., 2016; Liu Y et al., 2016; Melotti A et al., 2014; Sharmeen S et al., 2010; Draganov D et al., 2015), but the underlying mechanism was still uncleared. In this study, we observed that IVM induced cell death accompanied by sequential changes of the ER structure, including reorganization, vacuolation, and permeabilization of the ER. When we observed YFP-ER cells and Sec61-GFP cells stained with ER-Tracker Blue-White DPX by confocal microscopy, both YFP-ER and Sec61-GFP showed reticular ER structure in untreated cells without the fluorescence of ER-Tracker White DPX. ER-Tracker Blue-White DPX, a cell permeable dye that selectively labels the ER possibly due to its hydrophobic nature to make them compatible for partitioning into the ER membrane environment (Wijesooriya CS et al., 2019; Cloe L et al., 2000), and ER-Tracker Blue-White DPX is preferentially attached to the thiol groups
(Nandi S et al., 2018). And, in our data. ER-Tracker Blue-White DPX was colocalized Sec61-GFP fluorescence. These results suggested that ER-Tracker Blue-White DPX indicated the ER membrane. In both YFP-ER and Sec61-GFP cells treated with IVM for 4 h, green or yellow fluorescence of round shape was exactly colocalized with ER-Tracker Blue-White DPX. In addition, EM showed the stacked membrane structure of round shape around the nuclei at this time. These ER structures were different from those with the fluorescence of YFP-ER-(+) inside of the ER-derived vacuoles and Sec61-(+) around the derived vacuoles at 8 h of IVM treatment. In addition, ER-Tracker Blue-White DPX was detected only outside of the ER vacuoles. These
- 54 -
results suggest that IVM-induced ER membrane stacking or aggregation precedes the ER-derived vacuolation. Very interestingly, from 16 h of IVM treatment, phase-contrast microscopy showed that ER-derived vacuoles, which were evidently detected at 8 h of IVM treatment, disappeared. The fluorescence of YFP-ER cells was diffusively scattered in the cellular space including the cytosol and nuclei, except the cellular regions, which were presumed to be megamitochondria. Structurally, the ER is a network of membranes found throughout the cells and connected to the nucleus. Since rough ER is attached to the nuclear envelope that surrounds the nucleus (Dultz E et al., 2007; Gia K. Voeltz et al., 2002), nuclear membrane integrity also may be also disrupted, when the ER membrane lose its integrity. As a result, ER luminal protein might be diffused also into the nuclei at late phase of IVM treatment. The fluorescence of Sec61-GFP reappeared with less stacked patterns in the cytosol and was exactly co-localized with that of ER-Tracker Blue-White DPX. These results suggest that the boundaries of the ER-derived vacuoles and the nuclei are disrupted and the squeezed and stacked ER membrane structures lost the tension due the permeabilization of the ER. In ER permeabilization, Sec61-GFP and ER-tracker were co-localized in re-formed ER reorganization structure. But Its size was larger and less compacted than the ER reorganization.
ER reorganization is a structure characterized by aggregated or stacked ER membrane (Erik L. Snapp et al., 2003). Previous studies reported that ER reorganization was induced by imbalance of ion homeostasis (Ca2+, Na+), overexpression of ER membrane protein, disruption of vesicle trafficking (Erik L. Snapp et al., 2003; Li X et al., 2016; Varadarajan S et al., 2012; Varadarajan S et al., 2013; Korkhov VM et al., 2009). Among these stimuli
- 55 -
of ER reorganization, imbalance of ion homeostasis is also reported as the stimulus of ER vacuolation (Yoon MJ et al., 2012; Yoon MJ et al., 2014). Previously, tunicamycin, an ER stress inducer, was reported to induce ER reorganization, and after induction of ER reorganization, UPR signals were activated (Varadarajan S et al., 2012). ER stress is one of the causes to induce ER vacuolation (Lee D et al., 2016). Therefore, there may exist some extent of correlation between ER reorganization and ER vacuolation, although further detailed study is required. In this study, we observed vesicle-like lipid droplet in electron microscopy image. Therefore, we examined whether the formation of lipid droplets is associated with IVM-induced changes in the ER structure. We found that addition of oleic acid, an inducer of lipid droplet (Lucía C. Lagrutta et al., 2017), promoted IVM-induced ER reorganization and pretreatment with triacsin C, an inhibitor of acyl-CoA synthetase which is known to inhibit the formation of lipid droplets (Dechandt CRP et al., 2017), effectively reduced it. However, either oleic acid or triacsin C, did not affect IVM-induced vacuolation and permeabilization of the ER and subsequent cell death. These results suggest the possibility that IVM-induced ER reorganization is closely linked to lipid homeostasis, but not to other changes in the ER structure and cell death.
There are two basic types of ER, such as rough ER and smooth ER (Schwarz DS et al., 2016). Rough ER is called rough because it has ribosomes attached to its surface and it look like sheets or disks of bumpy membranes. In contrast, smooth ER looks like tubes and important in the creation and storage of lipids and steroids (Gia K. Voeltz et al., 2002). Since ER reorganization is known to be derived from smooth ER and IVM increases lipid droplets at early phase and then induces ER reorganization, suggesting that disruption of lipid
- 56 -
homeostasis related to smooth ER may be critically involved in IVM-induced ER reorganization.
The ER is the central organelle for protein translocation, protein folding, and protein post-translational modifications (Sano R et al., 2013). When the ER undergoes excessive workload over the capacity of protein folding, ER stress is induced, and to cope with this stress, UPR signaling pathway is activated. In the present study, IVM increased the expression of ER stress-related proteins.
ER stress is one of the causes of paraptosis, because many paraptosis inducers accumulates misfolded proteins and blocking of protein synthesis inhibits the induction of paraptosis (Sperandio S et al., 2004; Lee D et al.,
2016). In addition, ER vacuolation is a key morphological feature of paraptosis. Since we found that IVM induces ER vacuolation prior to cell death, we investigated whether IVM-induced cell death is associated paraptosis. When we pretreated MDA-MB 435S cells with CHX, a protein synthesis inhibitor, IVM-induced ER stress, ER vacuolation, ER permeabilization, and subsequent cell death. These results suggest that the increase of ER stress play a critical role in IVM induced paraptosis. Interestingly, IVM-induced ER reorganization was rather increased by pretreatment of CHX. Since CHX was previously shown to increase the levels of cholesterol esters (Suzuki M et al., 2012), a source of lipid droplet, CHX-mediated further increase in ER reorganization in IVM-treated cancer cells may be due to the increase in cholesterol levels. These results suggest that disruption of protein homeostasis more critically contribute to IVM-induced cell death than the disruption of lipid homeostasis.
- 57 -
In addition, paraptosis was shown to be mediated by the mitogen-activated protein kinase (MAPK), such as MEK-2 or JNK (Sperandio S et al., 2004). In our data, IVM activated MAPKs, such as JNK, ERK, and p38. Pretreatment with U0126, a specific inhibitor of MEK, and SB203580, a specific inhibitor of p38, delayed IVM-induced vacuolation and permeabilization of the ER and subsequent cell death. Collectively, these results suggest that ER stress and MAPK pathway are positively involved in IVM-induced cell death and IVM may induce paraptosis in MDA-MB 435S cells.
ER stress can be induced also by imbalance of Ca2+ homeostasis, disruption
of thiol homeostasis, and increased intracellular ROS (Delic et al., 2012; Ellgaard et al., 2001; Yoon MJ et al., 2010; Ghosh K et al., 2016). When we tested the possible involvement of Ca2+ in IVM-induced paraptosis, IVM
increased intracellular Ca2+ homeostasis, and IVM-induced vacuolation and
permeabilization of the ER, and cell death was inhibited by pretreatment of BAPTA-AM, an intracellular Ca2+ chelator. These results suggest that
disruption of Ca2+ homeostasis play a crucial role in IVM-induced paraptosis.
Interestingly, pretreatment with BAPTA-AM did not affect IVM-induced ER stress responses, except CHOP induction, whereas CHX effectively blocked IVM-induced increase of intracellular Ca2+ levels. Therefore, these results
suggest that Ca2+ imbalance may not be a main cause of IVM-induced ER stress or Ca2+ levels may be increased downstream of IVM-induced ER stress. IVM is known to kill the parasites via induction of membrane hyperpolarization due to irreversible opening of Cl- channels such as
γ-aminobutyric acid (GABA) receptors or glutamate-gated chloride