의학
의학
의학
의학 박사학위
박사학위
박사학위
박사학위 논문
논문
논문
논문
Knockdown of nfa1 Gene Cloned from
Naegleria fowleri by Antisense RNA
아
아
아
아 주
주
주 대
주
대
대 학
대
학
학
학 교
교 대
교
교
대
대
대 학
학
학
학 원
원
원
원
의
의
의
의 학
학
학 과
학
과
과
과
이
이
이
이 상
상
상 철
상
철
철
철
Knockdown of nfa1 Gene Cloned from
Naegleria fowleri by Antisense RNA
by
Sang-Chul Lee
A Dissertation Submitted to The Graduate School of Ajou University
in Partial Fulfillment of the Requirements for the Degree of
DOCTOR OF PHILOSOPHY
Supervised by
Ho-Joon Shin, Ph.D.
Department of Medical Sciences
The Graduate School, Ajou University
이상철
이상철
이상철
이상철의
의
의
의 의학
의학
의학
의학 박사학위
박사학위
박사학위
박사학위 논문을
논문을
논문을
논문을 인준함
인준함
인준함.
인준함
심사
심사
심사
심사 위원장
위원장
위원장 이
위원장
이
이 일
이
일
일
일 영
영
영
영 인
인
인
인
심
심
심
심 사
사
사 위
사
위
위
위 원
원
원 신
원
신 호
신
신
호
호
호 준
준
준
준 인
인
인
인
심
심
심
심 사
사
사
사 위
위
위
위 원
원 박
원
원
박
박 선
박
선
선
선 인
인
인
인
심
심
심
심 사
사
사 위
사
위
위
위 원
원
원 김
원
김 경
김
김
경
경
경 민
민
민
민 인
인
인
인
심
심
심
심 사
사
사 위
사
위
위
위 원
원
원 임
원
임 경
임
임
경
경
경 일
일
일
일 인
인
인
인
아
아
아
아 주
주
주
주 대
대
대
대 학
학
학
학 교
교 대
교
교
대
대
대 학
학
학
학 원
원
원
원
2005년
년
년
년 12월
월
월
월 22일
일
일
일
-ABSTRACT-
Knockdown of nfa1 Gene Cloned from
Naegleria fowleri by Antisense RNA
PAME caused by N. fowleri is an acute, fulminant, and rapidly progressing fatal illness that usually affects children and young adults. There have been few reports to address proteins functioning in vitro cytotoxicity of N. fowleri. The nfa1 gene, cloned from a cDNA library of N. fowleri by immunoscreening, should be concerned with the formation of food-cups that is a phagocytic structure. In addition, an anti-Nfa1 antobody reduced the in vitro cytotoxicity of N. fowleri against target cells. To elucidate the function of proteins cloned from N. fowleri, the gene-knockdown analysis by a transfection system are not yet established. In this present study, to describe the association of an Nfa1 protein in vitro cytotoxicity of N. fowleri to target cells, an antisense RNA or siRNA of nfa1 gene were transfected into N. fowleri trophozoites. By the synthetic dsRNA of nfa1 gene ORF, the expression of nfa1 gene and the Nfa1 protein were knockdowned about 50% and 30%, respectively. However, by antisense RNA transcribed in vitro, the expression of nfa1 gene and the Nfa1 protein were less knockdowned than those of dsRNA of nfa1 gene. Four synthetic siRNAs were not act equally, but a sinfa1-1 was highly effective to knockdown the
nfa1 gene and Nfa1 protein with 70% and 43%, respectively. However, N. fowleri
vitro cytotoxicity against murine macrophages as compared with normal N. fowleri trophozoites. Therefore, a vector-based system, in which transfected genes can be maintain longer, was used to transfect the nfa1 gene into N. fowleri. A pAct/SAGAH vector with a sinfa1-1 and a pAct/asnfa1AGAH vector with an asRNA of the nfa1 gene ORF were cloned, and then transfected into N. fowleri. By the pAct/SAGAH vector, the expression of nfa1 gene and the Nfa1 protein were knockdowned as 60% and 29%, in comparison with the pAct/asnfa1AGAH vector of 30% in the nfa1 gene and 18% in Nfa1 protein. In particular, the in vitro cytotoxicity of N. fowleri transfected with a pAct/SAGAH vector against macrophages was decreased to 26.6% at 17 h and 26.8% at 24 h post co-incubation, whereas the in vitro cytotoxicity was decreased to 7.4% at 17 h and 6.6% at 24 h by a pAct/asnfa1AGAH vector. These results suggest that the function of RNAi should be worked in N. fowleri trophozoites. Therefore, single stranded RNA, dsRNA, siRNA, and siRNA-vector were not only efficiently transfected into N. fowleri using each transfection reagent, but also decreased the function of Nfa1 protein which plays very important role in destroying macrophages. This result may be helpful for understanding the function of Nfa1 protein as a target cell-cantact mechanism in the N. fowleri infection.
____________________________________________________________________ Key Words: N. fowleri, PAME, nfa1 gene, RNAi, cytotoxicity
TABLE OF CONTENTS
ABSTRACT --- i
TABLE OF CONTENTS --- iii
LIST OF FIGURES --- v
LIST OF TABLES --- vii
I. INTRODUCTION --- 1
A. Free-living amoeba Naegleria fowleri --- 1
B. Life cycles and occurrence of PAME by N. fowleri --- 2
C. Pathogenic factors related to PAME by N. fowleri --- 5
D. The nfa1 gene --- 6
E. Antisense RNA regulation and RNA interference --- 7
1. Antisense RNA regulation --- 7
1-1. Antisense RNA regulated system in eukaryotes --- 8
2. RNA interference --- 9
2-1. Mechanism of RNAi --- 11
2-2. Delivery, practical aspects and problems of RNAi --- 13
F. Subjects in this study --- 15
II. MATERIALS AND METHODS --- 16
A. Cultivation of N. fowleri trophozoites and murine macrophages --- 16
B. Total RNA preparation and RT-PCR --- 16
C. Synthesis of antisense- and sense-directed RNA of nfa1 gene ORF, and siRNAs of the nfa1 gene --- 17
D. Southern blot hybridization --- 19
E. Transfection of asnfa1, dsnfa1 and siRNAs into N. fowleri --- 20
F. Northern blot hybridization and quantitation of RNA --- 20
G. Cell lysate preparation and immunoblots --- 21
H. Vector construction of sinfa1-1 and asnfa1 --- 22
I. Transfection of pAct/sinfa1-1AGAH and pAct/asnfa1AGAH vector into N. fowleri trophozoites and hygromycin selection --- 24
J. Indirect immunofluorescence antibody test --- 26
K. In vitro cytotoxicity --- 27
III. RESULTS --- 28
A. In vitro synthesis of ssnfa1, asnfa1 and dsnfa1 --- 28
B. Knockdown of the nfa1 mRNA and Nfa1 protein by asnfa1 or dsnfa1 -- 31
C. Knockdown of the nfa1 mRNA and Nfa1 protein by synthetic siRNAs -- 35
D. RNAi function by a vector-based system --- 40
E. Expression of an Nfa1 protein in N. fowleri transfected with a RNAi vector --- 45
F. In vitro cytotoxicity of N. fowleri transfected with a RNAi vector --- 49
IV. DISCUSSION --- 51
V. CONCLUSION --- 60
REFERENCES --- 62
LIST OF FIGURES
Fig. 1. Life cycles of N. fowleri and PAME --- 3 Fig. 2. Model for RNAi --- 11 Fig. 3. Schematic representation of an nfa1 gene in N. fowleri used to produce sense (ss) and antisense (as) RNA followed by dsRNA formation
in vitro --- 29 Fig. 4. Findings of dsnfa1 synthesis and Southern blotting --- 30 Fig. 5. Northern blotting and quantitative analysis of the nfa1 gene mRNA
from N. fowleri trophozoites transfected with an asnfa1 or dsnfa1 --- 33 Fig. 6. Western blotting and quantitative analysis of the Nfa1 protein
from N. fowleri trophozoites transfected with an asnfa1 or dsnfa1 --- 34 Fig. 7. GFP fluorescence in N. fowleri transfected with GFP-conjugated
with siRNA of Lamin A/C gene --- 36 Fig. 8. Northern blotting and quantitative analysis of the nfa1 gene mRNA
from N. fowleri trophozoites transfected with the siRNAs
of an nfa1 gene --- 38 Fig. 9. Western blotting and quantitative analysis of the Nfa1 protein from
N. fowleri trophozoites transfected with the siRNAs of an nfa1 gene -- 39
Fig. 10. Vector construction for RNAi in N. fowleri --- 41 Fig. 11. Feasibility of transfection reagents for transfection into N. fowleri
Fig. 12. Northern blotting and quantitative analysis of the nfa1 gene mRNA
from N. fowleri trophozoites transfected with the RNAi vector --- 44 Fig. 13. Western blotting and quantitative analysis of the Nfa1 protein
from N. fowleri trophozoites transfected with the RNAi --- 46 Fig. 14. The fluorescence of the Nfa1 protein by immunocytochemistry --- 48
LIST OF TABLES
I. INTRODUCTION
A. Free-living amoeba Naegleria fowleri
Naegleria fowleri is the causal agent of primary amoebic meningoencephalitis
(PAME) which is fulminant and rapidly fatal disease that affects mainly children and young adults (Carter, 1970). Naegleria spp. are amoeboflagellates found in soil and water, but they are not as ubiquitous as Acanthamoeba, by which granulomatous amoebic encephalitis (GAE) is occurred as a subacute or chronic disease with focal granulomatous lesions in the brain. In a swimming pool that had been identified as the source of an outbreak of 16 PAME cases, N. fowleri and other thermophilic
Naegleria spp. were found to proliferate in a cavity behind a damaged wall of the
pool (Kadlec et al., 1980). Although 30 species of Naegleria have been recognized based upon sequencing data (De Jonckheere, 2004), N. fowleri is the only one that has been isolated from cases of PAME. Other species (N. australiensis, N. italica, N.
pilippinensis) may be pathogenic in the mouse model of PAME, but have not been
identified from any human cases. Because, it grows at somewhat elevated temperature, the amoeba has been isolated from warm-water bodies including man-made lakes and ponds, hot spring, and thermally polluted streams and rivers. But thermotolerance is not the sole factor determining pathogenicity of Naegleria spp. but is non-pathogenic in the mouse model for PAME (De Jonckheere et al., 1977,
Stevens et al., 1980)
B. Life cycles and occurrence of PAME by N. fowleri
The Naegleria life cycle includes amoeboid and cystic stages, as well as a flagellate stage which develops from the amoeba (Fig. 1). The trophic amoeba has distinctive limacine (slug-like) pattern of locomotion, with one or more ectoplasmic psuedopods. When the amoeba undergoes a morphogenetic transformation (over 30−60 min) and triggered by suspension in water, trophozoites are changed into a transitory non-feeding and non-dividing flagellate stage. The cyst stage has a double wall with pores. All Naegleria spp. are morphologically similar, if not identical to one another, although some differences among species may be found, as in cyst pore structure (Pussard and Pons, 1979). In nature and in the laboratory, the amoeba feeds actively on bacteria. Isolations of N. fowleri from environmental soil or water samples are accomplished by growth on nutrient agar plates coated with Escherichia
coli at 45℃ (Lares-Villa et al., 1993). Other bacteria (preferably non-mucoid strains),
such as Enterobacter aerogenes or Klebsiella pneumoniae, may also be used. PAME is a fulminating disease, developing within several days following exposure to the water source, and causing death within 1−2 weeks after hospitalization (Fig. 1).
Fig. 1. Life cycles of N. fowleri and PAME. The distinctive feature of N. fowleri
trophozoite is the presence of multiple finger-like projections, pseudopodia. Trophozoite or flagellate form can infect human and experimental mouse via olfactory bulb, resulting in PAME.
Olfactory nerve Epithelium Brain
PAME
Trophozoite Flagellate Cyst Trophozoite PseudopodiumBecause of the rapid onset of the infection and the delay in diagnosis, few individuals survive. Infection results from introduction of water containing amoebae into the nasal cavity and subsequent passage of the organisms to the central nervous system (CNS) via the olfactory apparatus (Carter, 1968, 1970, 1972). Acute hemorrhagic necrotizing meningoencephalitis follows invasion of the CNS. Only amoebic trophozoites are found in the lesions of patients with PAME (Fig. 1). PAME is characterized by the sudden onset of bifrontal or bitemporal headache, fever (from 38.2℃ to >40℃), nausea, vomiting (usually projectile), signs of meningeal irritation, and encephalitis. There is often a rapid progression from fever and early signs of leptomeningitis, encephalitis, or meningoencephalitis to coma and seizures. Nausea, vomiting, photophobia, and other symptoms related to increase intracranial pressure may also be prominent. The final diagnosis of PAME is based on the isolation and culture of free-living amoebae from cerebrospinal fluid (CSF) or the demonstration of amoebic trophozoites in biopsied brain tissue. Clinical diagnosis by experienced practitioners is based on the characteristic stromal infiltrate. Antibodies may be detected in serum; however, serologic tests usually are of no value in the diagnosis of infections with free-living amoebae (John, 1982). Amphotercin B reportedly cured one case of PAME (Ma et al., 1990). High morbidity and mortality may be reduced with rapid diagnosis and earlier treatment. A drawback in the use of Amphotericin B is its potential for impairment of kidney function. This has stimulated testing of other less toxic forms of the drug for use in treatment. Amphotericin B methyl ester is one of the less damaging forms of the compound, but it is less effective in treatment in
the mouse model of PAME (Ferrante, 1982). Other antimicrobials that have been tested, mostly in vitro, are clotrimazole, itraconazole, fluconazole, and ketoconazole, with varying degrees of efficacy. Differences in reported drug sensitivity are due to the use of different N. fowleri strains in different laboratories, which show variation to drugs. Amphotericin B, however, remains the drug of choice for PAME treatment.
C. Pathogenic factors related to PAME by N. fowleri
It is still unclear that the molecular mechanisms of invasion and pathogenesis of PAME by N. fowleri are still unclear. The pathogenicity would be a complex process which involves both contact-dependent and contact-independent pathways in order to kill host cells quickly and to reduce the degree to which defense can be induced. However, in general, adhesion is one of the crucial steps for the pathogenicity of amoeba as non-pathogenic amoeba exhibit significant decreased binding to host cells (Yang et al., 1997). The ability of N. fowleri to produce such a rapidly fatal infection has encouraged to search for virulence properties of the amoeba that might serve as virulence factors are the release of the enzymes phopholipase (Cursons et al., 1978), or neuraminidase (Eisen and Franson, 1987), the secretion of cystein proteinase (Aldape et al., 1994), the creation of pores in target cell membranes that may have a lytic effect (Young and Lowrey, 1989; Herbst et al., 2002) and aggressive phagocytotic activity (Brown, 1979). The amoeboid stage forms food cups (amoebostomes) that are capable of pinching off bits of target cell cytoplasm (Brown,
1979; John et al., 1985, Kang et al., 2005). In particular, as related with contact-dependent cell killing by N. fowleri, a few genes have been cloned and characterized in our previous studies (Cho et al., 2003; Shin et al., 2001).
D. The nfa1 gene of N. fowleri
An antigen-related gene was cloned from cDNA expression library of N. fowleri by immunoscreening with sera obtained from mice that were either immunized with an amoebic lysate or infected with trophozoites. The coding sequence of the cloned gene consisted of 357 bases that were translated into 119 amino acids. This gene was designated as nfa1 gene (N. fowleri antigen 1). The amino acid sequence of Nfa1 protein shares 43% identity, especially 100% in conserved regions and iron-binding residues, with the myohemerythrin protein of a marine annelid, Nereis diversicolor (Takagi et al., 1991; Shin et al., 2001). In our previous study, the Nfa1 protein was observed to localize specifically in pseudopodia using an anti-Nfa1 polyclonal antibody by a transmission electron microscopy (Shin et al., 2001). More recently, when N. fowleri trophozoites were cultured with CHO target cells, the Nfa1 protein was observed to localize in food-cups of phagocytic organelles (Kang et al., 2005). Anti-Nfa1 monoclonal antibodies were also produced using hybridoma (unpublished yet). When an anti-Nfa1 monoclonal antibody was used to observe the location of the Nfa1 protein in N. fowleri trophzoites, the Nfa1 protein was localized in pseudopodia. Because the location of the Nfa1 protein by the anti-Nfa1 polyclonal antibody was
identical with that by the anti-Nfa1 monoclonal antibody, and the Nfa1 protein was not detected in Acanthameobae by the anti-Nfa1 polyclonal antibody (Cho et al., 2003), it was suggested that the anti-Nfa1 polyclonal antibody be a mono-specific antibody. N. fowleri trophozoites showed the cytopathic effect on CHO target cells in a co-culture systemd. Otherwise, CHO cells co-cultured with N. fowleri trophozoites and an anti-Nfa1 antibody showed less destruction in a dose-dependent manner (Jeong et al., 2004). When the nfa1 gene from N. fowleri was tranfected into non-pathogenic N. gruberi, the in vitro cytotoxicity of N. gruberi was slightly increased (Jeong et al., 2005).
E. Antisense RNA regulation and RNA interference
1. Antisense RNA regulation
The first natural antisense RNAs were discovered in 1981 independently in Tomizawas and in Nordströms laboratories. These authors found that small plasmid-encoded RNA regulators control the copy numbers of the E. coli plasmids ColE1 and R1, respectively (Stougaard et al., 1981; Tomizawas et al., 1981). Antisense RNAs are small, diffusible, highly structured RNAs that act via sequence complementarity on target RNAs called sense RNAs. In eukaryotes, some processes like splicing or editing make use also of complementary small RNAs; however, these RNAs are not independent regulators, and therefore not regarded as bona fide antisense RNAs. In
the classical case, antisense RNAs are encoded in cis, i.e. they are transcribed from a promoter located on the opposite strand of the same DNA molecule, and therefore fully complementary to their target RNAs. However, over the past years, a number of antisense RNAs were detected that are encoded in trans, reveal only partial complementarity to their target RNA and have more than one target. The sense RNAs are mostly mRNAs encoding proteins of important/essential functions. In the majority of cases, antisense-RNA action entails posttranscriptional inhibition of target RNA function. However, a few activating antisense RNAs have been found, too. Some antisense RNAs (those involved in plasmid copy number control and postsegregational killing) are unstable, others (most chromosomally encoded and a few phage and transposon antisense RNAs) are stable.
1-1. Antisense RNA regulated system in eukaryotes
Eukaryotic antisense RNAs have been found only accidentally, and in most cases, their regulatory roles and the mechanism of action are still elusive. They seem to act preferentially via destabilization of the sense RNAs, but inhibition of splicing or translation has been suggested, too. Destabilization has been attributed to targeting of the antisense/sense-RNA duplex to dsRNase. Three cases are well studied. Antisense-RNA-mediated mRNA destabilization occurs mostly in the cytoplasm (Hildebrandt and Nellen, 1992). In mammalian cells, the stability of the eIF2α-mRNA is regulated by a differentially expressed antisense transcript originating from a promoter located
in the first intron of the eIF2α gene. In plants, no naturally occurring antisense RNAs have been found so far. However, artificially introduced antisense transcripts are believed to target mRNA for degradation. Short antisense RNAs can be also generated by RNA-dependent RNA polymerase (RdRp) from aberrant sense RNAs. In addition to RNA interference (RNAi), such RNAs can mediate methylation of homologous DNA sequences in the plant genome, thus silencing gene expression, a pathway which might be used by natural RNA regulators as well (Bender 2001; Matzke et al., 2001). Recently, a large number of small RNAs with probable regulatory functions have been discovered in Caenorhabditis elegans (Lau et al., 2001; Lee and Ambros, 2001). The expression of some of these microRNAs (miRNAs) varies during larval development, and the potential orthologs of several of these miRNA genes were identified in Drosophila and human genomes. These findings indicate that small regulatory RNAs may be ubiquitous in eukaryotes, too.
2. RNA interference
RNA interference (RNAi) is the induction of sequence-specific gene silencing by double-stranded RNA (dsRNA) (Fig. 2). It occurs posttranscriptionally and involves mRNA degradation. The term RNAi was coined after the discovery that the injection of double-stranded RNA (dsRNA) into C. elegans interferes with the expression of specific genes highly homologous in sequence to the delivered dsRNA (Fire et al., 1998).
RNAi is related to the "posttranscriptional gene silencing" (PTGS) or "cosuppression" phenomena observed in plants (Baulcombe, 1999; Vaucheret et al., 2000; Waterhouse et al., 1999) and "quelling" (silencing of an endogenous gene by the introduction of a transgenic copy of the gene) observed in Neurospora (Cogoni and Maciano, 1999, 1999). RNAi is a powerful tool that makes gene inactivation possible in organisms that were not amenable to genetic analysis before. In nature, RNAi may both play an important biological role in protecting the genome against instabilities caused by transposons and repetitive sequences (Ketting et al., 1999; Tabara et al., 1999) and be an ancient antiviral response/protection mechanism in both animals and plants (Baulcombe, 1999, Grant, 1999; Ratcliff et al., 1999).
2-1. Mechanism of RNAi
Biochemical analysis of RNAi became possible with the development of an in vitro Drosophila embryo lysate system for dsRNA-dependent gene silencing (Tuschl et al., 1999). A key finding was that small (21–23 nucleotides) dsRNAs called short interfering RNAs (siRNAs) are generated from the input dsRNA during PTGS and RNAi (Hammond et al., 2001). These small RNAs have been detected in plants,
Drosophila and C. elegans and have been suggested to serve as guide RNAs for
target recognition. In Drosophila extracts, these siRNAs with their 3′-OH and 5′-phosphate termini resemble breakdown products of an RNase III-like enzyme, e.g.,
Fig. 2. Model for RNAi. Antisense RNA strands are drawn in thin black, sense RNA
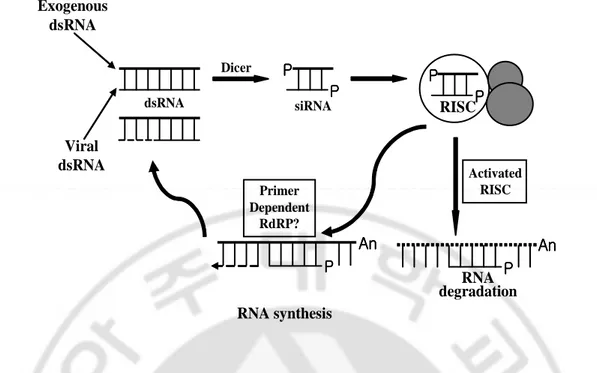
strands in thick black. Sense target RNA is shown in spotted lines, antisense target RNA in short thin line. The dsRNA processing proteins containing an RNA binding domain and a dsRNA-specific endonuclease domain are illustrated as grey-colored small and big ovals. The protein domain of small oval binds in the 5′–3′ direction, the protein domain of big oval in the 3′–5′ direction. Only the siRNA associated with the domain of big oval is able to guide target RNA cleavage. The RNA-induced silencing complex (RISC) is shown as large oval. A conformational change is proposed to occur in the RISC before target RNA cleavage because the cleavage site of the target RNA is displaced by 10–12 nt relative to the dsRNA processing site. The cleaved target RNA is directed into the processing pathway where it will be sequentially degraded. This figure was based on Elbashir et al (2001).
P P P P P P P P Dicer P P P P P P P P RISC P P P P An AnAn An P PP P An An An An Primer Dependent RdRP? dsRNA Activated RISC RNA synthesis RNA degradation siRNA Exogenous dsRNA Viral dsRNA
Dicer, digestion (Elbashir et al., 2001). Synthetic siRNAs can also induce gene-specific inhibition of expression in Drosophila extracts (Tuschl et al., 1999), in insect and mammalian cell lines (Elbashir et al., 2001; Caplen et al., 2001) and in C.
elegans indicating that the dsRNA-processing step and the targeting step can be
uncoupled. In each case, the interference was superior to the inhibition mediated by single-stranded antisense oligonucleotides (Bass, 2000). A model tries to explain why the double-stranded trigger RNA is cleaved into small fragments. Cleavage to segments of 21–23 nt might provide optimal specificity for a homology-based searching mechanism. Much shorter segments would leave insufficient specificity, while much longer segments might allow unwanted attacks on cellular genes with partial but extended identity to the trigger.
The following model for dsRNA-directed mRNA cleavage is proposed (based on (Bass, 2000; Berstein et al., 2001; Elbashir et al., 2001). The dsRNA is cleaved to 21–23-nt-long fragments by Dicer or a Dicer homologue. Processing starts from the ends of the blunt-ended dsRNA or dsRNAs with short 3′ overhangs and proceeds in 21–23-nt steps. The resulting fragments (siRNAs) are bound by RNAi-specific enzymes possibly still including Dicer and could be incorporated into a distinct nuclease complex (RNA-induced silencing complex; RISC) that targets mRNA for degradation. In this complex, they pair with the target mRNA and cleave the mRNA in the center of the region recognized by the siRNA whereby the mRNA cleavage boundaries are determined by the sequence of the dsRNA. Either the same RNase that cleaves the dsRNA or another RNase that has to be recruited cleaves the target
RNA, probably by temporarily displacing the passive siRNA strand not used for target recognition. The dsRNA-processing proteins or a subset of them remains associated with the siRNA duplex after the processing reaction. The orientation of the siRNA duplex relative to these proteins determines which of the two complementary strands functions in guiding target RNA degradation. Chemically synthesized siRNA duplexes guide cleavage of sense as well as antisense target RNA, as they are able to associate with protein components in either possible orientation. Adenosine triphosphate (ATP) may be required for complex formation on the dsRNA, strand dissociation during or after dsRNA cleavage, pairing of the 21–23-nt RNAs with the target mRNA, mRNA cleavage and recycling of the targeting complex. Therefore, an RNA-dependent ATPase, or RNA helicase, is probably associated with the RISC.
2-2. Delivery, practical aspects and problems of RNAi
For dsRNA delivery, several methods can be applied. Electroporation is used in simpler organisms, whereas microinjection of dsRNA into germ line or early embryo is the method of choice in multicellular organisms. In C. elegans, injection into the intestine or pseudocoelom is almost as efficient as injection into the germ line. Even feeding worms with bacteria that express dsRNA, or soaking worms in dsRNA solutions has been applied with success (Tabara et al., 1998; Tabara et al., 1999; Timmons and Fire, 1998). Because RNAi is homology-dependent, single base pair
mismatches between siRNA and target RNA dramatically reduce silencing. The length of the dsRNA can affect the RNAi efficiency (Bosher et al., 1999). Usually, dsRNA of at least 500 bp is applied but recently it has been found that perfectly matching duplexes as short as 21 bp suffice (Caplen et al., 2001; Parrish et al., 2000).
When dsRNA is injected into early embryos, it is diluted upon cell division. Therefore, early genes are more easily inactivated than late genes, which is especially a problem for higher organisms (in mouse, a construct was effective only until a 40–50-fold increase in cell mass) (Wianny and Zernicka-Goetz, 2000). In C.
elegans, the application of a plasmid with inducible (by heat-shock) promoter for the
production of dsRNA (sense RNA and antisense RNA expressed in the form of a hairpin) made inheritable RNAi possible (Tavernarakis et al., 2000). With this approach inheritable transgenes are easily generated, large numbers of mutant organisms can be propagated delivering enough material for a broad variety of analyses and stage-specific RNAi can be performed. Furthermore, neurons, normally partially resistant to exogenous supply of dsRNA, became RNAi sensitive upon plasmid-derived in vivo supply of dsRNA (Clemens, 1997). The expression of dsRNA under the control of tissue-specific promoters instead of inducible ones is also conceivable.
Although experiments to elucidate the underlying mechanism progress rapidly, we are still at the beginning of our understanding of the molecular processes responsible for RNAi and the breadth of its function in biology. In contrast, practical applications have already allowed rapid surveys of gene functions (Fraser et al.,
2000) and will possibly result in new therapeutical interventions.
F. Subjects in this study
A great advance in studying the function of genes and biology of free-living ameobae has been the use of transfection technology. In Acanthamoeba, the transient and stable transfection system had been established (Hu and Henney, 1997; Yin and Henney, 1997; Peng et al., 2005). Transfection of the pEGFP–C2 vector with an nfa1 gene into nonpathogenic N. gruberi has been tried in our previous study, and transgenic N. gruberi has been maintained for nine months after the treatment of G418 antibiotics as a selective marker (Jeong et al., 2005).
In this study, to observe the gene-knockdown of the Nfa1 protein in N. fowleri, antisense RNA and siRNA of the nfa1 gene were constructed, cloned in a transfection vector and transfected into trophozoites of N. fowleri. In addition, to describe the association of Nfa1 protein with the in vitro cytotoxicity of N. fowleri to a target cell, N. fowleri tracfected with vectors including an antisense RNA or siRNA of the nfa1 gene was co-cultured with murine macrophages.
II. MATERIALS AND METHODS
A. Cultivation of N. fowleri trophozoites and murine macrophages
Trophozoites of N. fowleri (Carter NF69 strain, ATCC No. 30215) were cultured under axenic conditions in Nelson’s medium (Willaert, 1971). Trophozoites of N.
fowleri in log phase of growth were used in all experiments. N. fowleri was tested
before use for its ability to produce PAME in experimental mouse and for its cytopathic activity on CHO target cells (Jeong et al., 2004). Murine macrophages (ATCC No. TIB-71) were cultured with Dulbecco’s Minimal Essential Medium (DMEM, GibcoBRL) containing 10% fetal bovine serum (FBS, GibcoBRL)
(complete DMEM) at 37℃ in 5% CO2 incubator (Ralph et al., 1977).
B. Total RNA preparation and RT-PCR
Total RNA was prepared using an isolation kit RNAzol B (TEL-TEST, Fiendswood, TX, USA) solution. After 500 µl of RNAzol B solution was mixed with
pellet of 1 × 105 N. fowleri trophozoites by pipetting, 10 µl of chloroform was added
to the mixture. It was incubated on ice for 5 min and centrifuged at 10,000 × g for 15 min at 4℃. The supernatant was transferred to a new 1.5 ml tube and reacted with 250 µl of isoamylalcohol. Incubated for 15 min at 4℃, it was centrifuged at 10,000 ×
g for 15 min at 4℃. The pellet was washed with 1 ml of 70% ice-cold ethanol once and dried at room temperature (RT). The total RNA was suspended with 10 µl of diethylpyrocarbonate (DEPC)-treated distilled water (DW) and stored at -70℃. RNA yield and quality were evaluated spectrophotometrically and by analytical gel electrophoresis according to Sagerström and Sive (1996). For reverse transcription- polymerase chain reaction (RT-PCR), we used a Superscript First Strand Synthesis System kit (Invitrogen) to generate cDNA from 5 µg of total RNA pretreated with 1
µg of DNase I (Sigma). Reverse transcription was performed according to the
manufacturer’s recommendations for first-strand synthesis using gene-specific primers. PCR fragments were amplified from cDNA by using Taq polymerase (Promega) and 32 cycles of 95℃ for 30 s, 55℃ for 1 min, and 72℃ for 1 min.
C. Synthesis of antisense and sense-directed RNA of nfa1 gene ORF, and siRNAs of the nfa1 gene
To transcript antisense-directed RNA of an nfa1 gene open reading frame (ORF) (asnfa1), the nfa1 gene was cloned into pGEM−4Z vector (Promega) containing T7 promoter behind multicloning sites (MCS) using the method of a cohesive ligation to construct a pGEM−4Z/asnfa1 vector. The pGEM−4Z/asnfa1 vector was digested with the restriction enzyme of Eco RI for 2 h and transcribed in vitro. Briefly, approximately 500 ng of prepared DNA was added to the transcription mixture containing 10 µl of 10 mM NTP mix (2.5 mM ATP, 2.5 mM CTP, 2.5 mM UTP, 2.5
mM GTP; Invitrogen), 2.5 µl of 10 mM DTT, 2.5 µl of 10× buffer, 40 U of RNase inhibitor (Invitrogen) and 30 U of T7 RNA polymerase (Promega). The mixture was incubated at 37℃ for 1 h and the reaction was stopped by phenol/chloroform extraction. The transcripts were precipitated using 3 M sodium acetate and dissolved in DEPC-treated water. For sense-directed RNA of an nfa1 gene ORF (ssnfa1), the
nfa1 gene was first cloned into pGEM−3Zf(+) vector (Promega) containing T7
promoter at the front of MCS using the method of a cohesive ligation to construct a pGEM−3Zf(+)/ssnfa1 vector. The pGEM−3Zf(+)/ssnfa1 vector was digested with the enzyme of Hind III and the sense-directed nfa1 gene was transcribed using T7 polymerase. The transcript was precipitated with 3 M sodium acetate mentioned above. To make double stranded RNA of nfa1 gene ORF (dsnfa1), equal amounts of
ssnfa1 and asnfa1 were mixed, heated to 65℃ and annealed by slow cooling (65℃,
55℃,45℃, 35℃ and 25℃ for respective 30 min) for 2 h 30 min. Individual RNAs
and double stranded RNA were analyzed on 1% agarose gel. The four short double-stranded nfa1 gene, four synthetic siRNAs; sinfa1-1, correspondence of nucleotides 340–360 of the nfa1 gene ORF (Genebank Accession No. AF230370), 5’-r(AAGTACAAGGGAGTGCTTTAA)d(TT); sinfa1-2, correspondence of nucleotides 82–102 of the nfa1 gene ORF, 5’-r(AAGCTCTTTGCTCTCATCAAT)d(TT); sinfa1-3, correspondence of nucleotides 252–272 of the nfa1 gene ORF, 5’-r(AAGATGCTTTGGGTTTGAAG)d(TT); sinfa1-4, correspondence of nucleotides 196–216 of the nfa1 gene ORF, 5’-r(AAGGTGAATTTCTCTGATTCT)d(TT), were synthesized (Qiagen) and resuspended in siRNA suspension buffer as supplied
by the manufacturer to final concentration of 20 µM solution. After the solution of
siRNA was heated to 90℃ for 1 min and then incubated at 37℃ for 1 h, it was stored
frozen at -20℃ prior to transfection.
D. Southern blot hybridization
To identify ssnfa1 or asnfa1 made by in vitro transcription, southern blot hybridization was performed. Samples loaded on agarose gel were prepared by PCR. For DNA of an nfa1 gene from pGEM–3Zf(+)/ssnfa1 vector, primers (5’-ATGGCCACTACTATTCCATCA-3’ and 5’-TTAAAGCACTCCCTTGTACTT-3’) were used. In our previous study, for DNA sequence unrelated with the nfa1 gene cloned into pEGFP−C2 vector (Clontech), primers (5’-ACAACATCGAGGACGGC-AGCGTGCAGCTCG-3’ and 5’-GTTTGGACAAACCACAACTAGAATGCAGTG-3’) were used (Jeong et al., 2004). After electrophoresis, the gels were subjected to short wave UV irradiation for 2 min and the DNA was denatured in alkaline solution (10 N NaOH, 5 M NaCl) for 45 min at RT, followed by neutralization for 45 min. DNA was transferred to Hybond N+ membrane (Amersham) by dry blotting overnight. The membrane was cross-linked using XL-1500 UV Crosslinker
(Spectrolinker Co.). For probe preparation, RNA probe labeled with α-32P-UTP was
produced by random priming of in vitro transcribed ssnfa1 or asnfa1. The probe was
hybridized overnight at 65℃ in hybridization buffer. The membrane was washed and
E. Transfection of asnfa1, dsnfa1 and siRNAs into N. fowleri trophozoites
To observe the effect of asnfa1, dsnfa1, and siRNAs of the nfa1 gene, each RNA was transfected into N. fowleri according to the procedure of an RNAi starter kit
(Qiagen). The day before transfection, 4 × 104 trophozoites were cultured in 24 well
cell culture plate (Nunc) in 0.5 ml of Nelson complete medium at 37℃. On the day of transfection, 1 µg each RNA in Nelson medium was diluted to give a final volume of 100 µl, and mixed by vortexing. For complex formation, 6 µl of RNAiFect reagent (Qiagen) was added and mixed. And then the samples were incubated for 15 min at RT to allow formation of transfection complexes. While complex formation was taking place, Nelson medium was gently aspirated from the plate. After 300 µl of fresh Nelson complete medium was added, the complexes were drop-wised onto the
N. fowleri trophozoites. To observe the transfection of siRNAs using RNAiFect
reagent, green fluorescent protein (GFP) fused siRNA directed against the human lamin A/C gene, which was unrelated with the nfa1 gene, was used in parallel. The transfection was observed under a fluorescent microscopy using standard FITC exitation/emission filters (488 nm/507 nm).
F. Northern blot hybridization and quantitation of RNA
min, and size fractionated under denaturing conditions on agarose gels containing formaldehyde. The RNA was transferred to a Hybond N+ membrane (Amersham) and hybridized under stringent conditions as described previously (Bracha et al., 1995). The membrane was cross-linked using XL-1500 UV Crosslinker (Spectrolinker). For an nfa1 gene-specific probe, the nfa1 gene was amplified with primers of 5’-ATGGCCACTACTATTCCATCA-3’ and
5’-TTAAAGCACTCCCT-TGTACTT-3’ and eluted. The probe randomly labeled with α-32P-dATP was prepared
from the appropriate PCR fragment. It was hybridized overnight at 65℃ in
hybridization buffer. The membrane was washed and then exposed to imaging plate for 2 days. After the bands on imaging plate were read by Image Reader (FLA-3000, Fujifilm, Japan), they were quantified by a densitometer program of Image Guage (LAS1000, Fujifilm, Japan).
G. Cell lysate preparation and immunoblots
Cultures (12 ml) of N. fowleri trophozoites were harvested and the cell pellets were suspended with 200 µl of phosphate-buffered saline (PBS, pH 7.4; 137 mM
NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4). Pellets were solubilized by
freezing-thawing method (Im and Shin, 2003) to obtain a lysate. Protein content in cell-free extracts was determined according to Bradford (Bradford, 1976) with protein assay dye reagent and bovine serum albumin for standard curve calibration (Ultrospec 3000, Pharmacia Biotech). The lysate was boiled in sodium dodecyl
sulfide (SDS) treatment buffer (125 mM Tris-HCl, 4% (w/v) SDS, 20% (v/v) glycerol, 3.1% (w/v) dithiothreitol, 0.001% (w/v) bromophenol blue) for 5 min. The lysate was resolved for 2 h at 100 V on 15% polyacrylamide gel (15 µg/lane) under reducing conditions in a Mini-Protean II Cell (Bio-Rad) according to the method of Laemmli (Laemmli et al., 1970). The lysate was transferred electrophoretically to polyvinylidene difluoride (PVDF) membrane (Hybond-P; Amersham Biosciences) with transfer buffer (25 mM Tris, 192 mM glycine, 20% (v/v) methanol; without adjusting the pH, it should be around 8.0 − 8.2) at 250 mA for 1 h 30 min. After the blot was incubated with 3% bovine serum albumin (BSA) in PBS, it was reacted with a suspension of anti-Nfa1 polyclonal antibody (1:100) and then subjected to interact with an horseradishperoxidase (HRP)-conjugated anti-mouse IgG (1:2,000)(Sigma) and developed by enhanced chemiluminescence. Detection was done for only HRP reaction by autoradiography. The amount was determined to quantify the Nfa1 protein expression by a densitometer (LAS1000, Fujifilm, Japan). The densities of protein bands were divided by the corresponding densities recognized by the anti-Nfa2 antibody to obtain a normalized protein ratio. The standard curve of proteins was used to demonstrate that signals were in the linear range for quantitative purposes.
H. Vector construction of sinfa1-1 and asnfa1
RNAT−U6.1/Hygro (6.5 kb, Genscript) vector. The pRNAT−U6.1/Hygro vector was designed for mammalian transfection. It carries a hygromycin resistance gene as the selectable marker, GFP marker (coral GFP, cGFP) under cytomegalovirus (CMV) promoter control used to track the transfection efficiency. It uses U6 promoter, which is a kind of human polymerase III promoter to effectively transcribe short RNA. An sinfa1-1 was cloned into the pRNAT−U6.1/Hygro vector to construct a pRNAT−U6.1/sinfa1-1/Hygro vector by the manufacturer of Genscript company. In our previous study, U6, CMV and simian virus (SV) 40 promoter transfected into N.
fowleri trophozoites did not transcribe siRNA, cGFP, and hygromycin resistance
gene, respectively. The fluorescence of GFP was not also observed in N. fowleri transfected with a pRNAT−U6.1/Hygro vector (data not shown). It needs to change other promoter functioning RNA transcription. Therefore, 5’ untranslated regions (UTR) (kindly presented by Prof. Lee at Yonsei University) as a promoter of an actin gene from nonpathogenic N. gruberi was used to transcribe genes mentioned above. For cloning of 5’ UTR of an actin gene, three promoters of U6, CMV, and SV40 were excised from the vector using restriction enzymes. To clone final pAct/SAGAH vector (Act and A, 5’ UTR of an actin gene; S, siRNA of an nfa1 gene; G, GFP; H, hygromycin resistance gene) used to transfect into N. fowleri trophozoites, 5’ UTR of an actin gene in pEGFP−N2 vector (Clontech) was amplified with primers containing restriction enzymes sites as follows; for cloning of 5’ UTR of an actin gene in the location of excised CMV, primer containing Hind III (5’-AGTCCTAAGCTTTTT-CCTTTTTTAGAAGCC-3’) and primer containing Nhe I
(5’-ACTAGGGCTA-GCTTTGTTGAGTGTTTGAG-3’); for cloning of 5’ UTR of an actin gene in the location of excised U6, primer containing Bgl II (5’-AGTCCTAGATCTTTTCCTT-TTTTTAGAAGC-3’) and primer containing Bam HI (5’-ACTATTGGATCCT-TTGTTGAGTGTTTGAG-3’); for cloning of 5’ UTR of an actin gene in the location of excised SV 40, primer containing Avr II (5’-AGTCCTCCTAGGTTTCCTTTT-TTAGAAGCC-3’) and primer containing Xma I (5’-ACTAGCCCGGGTT-TGTTGAGTGTTTGAG-3’). Each amplified fragment was ligated into the pRNAT−U6.1/Hygro vector excised with respective restriction enzymes to create pAct/SAGAH vector. To also observe the effect of asnfa1 by a vector-based system, an antisense DNA of the nfa1 gene was cloned into pAct/AGAH vector, in which an sinfa1-1 was eliminated, to create pAct/asnfa1AGAH vector. After the PCR fragment of antisense DNA of the nfa1 gene was amplified with the primers of AAGTCCAAGCTTATGGCCACTACTATT-3’ containing Hind III site and 5’-AAGTCCGGATCCTTAAAGCACTCCCTT-3’ containing Bam HI site, it was ligated into pAct/AGAH vector to create a pAct/asnfa1AGAH vector. All cloned vectors were sequenced with ABI Perkin Elmer 373A automated DNA sequencer (Applied Biosystems, Foster city, CA).
I. Transfection of pAct/sinfa1-1AGAH and pAct/asnfa1AGAH vector into N.
fowleri trophozoites and hygromycin selection
SuperFect transfection reagent (Qiagen GmbH, Germany). As our previously data, SuperFect reagent was successfully used in the nonpathogenic N. gruberi transfection study (Jeong et al., 2005). The branches of the reagent radiate from a central core and terminate at charged amino groups which can then interact with negatively charged phosphate groups of nucleic acids (Tang et al., 1996). Briefly, the
day before transfection, 4 × 104 trophozoites were cultured in 24 well cell culture
plate (Nunc) in 0.5 ml of Nelson incomplete medium without penicillin and
streptomycin (Gibco BRL, Gaithersburg, MD) at 37℃ overnight. On the day of
transfection, 1 µg plasmid DNA in Nelson incomplete medium without antibiotics was diluted to give a final volume of 60 µl, and mixed by vortexing. For complex formation, 5 µl of SuperFect reagent was added and mixed. And then the samples were incubated for 15 min at RT to allow formation of transfection complexes. While complex formation was taking place, Nelson medium was gently aspirated from the plate and N. fowleri trophozoites were washed once with 4 ml of 1 × PBS. After 350 µl of fresh Nelson complete medium was added, the complexes were drop-wised onto the N. fowleri trophozoites. The transfection was observed under a fluorescent microscopy using standard FITC exitation/emission filters (488 nm/507 nm). For the selection of transfected N. fowleri trophozoites, hygromycin antibiotics (100 µg/ml) (Gibco BRL, Gaithersburg, MD) was added to the 24 well plate 5 days after transfection. The lethal dose of untransfected N. fowleri to hygromycin antibiotics was determined that it was died after 1 week.
J. Indirect immunofluorescence antibody test
Immunocytochemistry was used to observe the knockdown of Nfa1 protein in N.
fowleri transfected with a pAct/SAGAH or pAct/asnfa1AGAH vector. N. fowleri
trophozoites were cultured on 24 well cell culture plate (Nunc A/S, Roskilde, Denmark) overnight. After the culture medium was discarded, the trophozoites were washed with 0.85% saline three times and were done with cold 0.85% saline at third
time. 200 µl of 10% formalin in 0.85% saline was added and the plate was incubated
at RT for 30 min. The trophozoites were washed with 0.85% saline three times,
added 200 µl of 1% NH4OH to render them permeable, and then incubated at RT for
5 min. The prefixed and permeablized trophozoites were incubated with an anti-Nfa1 polyclonal antibody (1:100 dilution containing 3% BSA) at RT overnight. After several washes with PBS containing Tween 20 (PBST), the trophozoites were treated with a tetramethyl rhodamine isocyanate (TRITC)-conjugated AffiniPure rabbit anti-mouse immunoglobulin G secondary antibody (Jackson ImmunoResearch Laboratories) (1:2,000 dilution with 3% BSA) at RT for 2 h and then washed with PBST. Trophozoites were analyzed by fluorescence microscopy using standard FITC excitation/emission filters (488 nm/507 nm).
K. In vitro cytotoxicity
This experiment, in 96 well cell culture plates, was performed using 5 × 104 murine
macrophages either alone or cocultured with (i) 5 × 104 trophozoites of N. fowleri,
(ii) 5 × 104 trophozoites of N. fowleri transfected with a pAct/AGAH vector as a
control vector, (iii) 5 × 104 trophozoites of N. fowleri transfected with a
pAct/SAGAH vector, (iv) 5 × 104 trophozoites of N. fowleri transfected with a
pAct/asnfa1AGAH vector, (v) 5 × 104 trophozoites of N. fowleri transfected with
only SuperFect reagent, (vi) 5 × 104 trophozoites of N. fowleri supplemented with
hygromycin antibiotics (100 µg/ml). The total volume per well was 200 µl of DMEM. Murine macrophages and trophozoites were observed under an inverted microscope at 24 h intervals. A lactate dehydrogenase (LDH) release assay was performed to quantify in vitro cytotoxicity. For the LDH assay, 50 µl of reacted supernatant in each well was transferred to a 96 well assay plate. After addition of 50 µl of reconstituted assay buffer from a CytoTox96 Non-radioactive Cytotoxicity Assay kit (Promega, Madison, WI), the plate was incubated for 30 min at RT, and then 50 µl of stop solution was added. The reactants were read at 490 nm using an enzyme-linked immunosorbent assay reader. The formula used to calculate the percent in vitro cytotoxicity was as follows: (sample value − control value) × 100 /(total LDH release − control value). Total LDH release was obtained from 50 ul of supernatant released
III. RESULTS
A. In vitro synthesis of ssnfa1, asnfa1 and dsnfa1
Prior to synthesize a dsnfa1 to knockdown an nfa1 gene and Nfa1 protein in N.
fowleri, ssnfa1 and asnfa1 were synthesized by PCR. An nfa1 gene ORF was cloned
into a pGEM−3Zf(+) and pGEM−4Z vector to create a pGEM−3Zf(+)/ssnfa1 and pGEM−4Z/asnfa1 vector, respectively (Fig. 3). The transcription was driven by the T7 promoter. After restriction enzymes digestion of Hind III for ssnfa1 and Eco RI for asnfa1, the restricted fragment was transcribed in vitro using T7 RNA polymerase. For dsnfa1, ssnfa1 and asnfa1 were equally mixed and annealed by slow-cooling method (Malhotra et al., 2002). Individual RNAs and double stranded RNAs were analyzed on 1% agarose gel (Fig. 4A). As the fragments were compared with each other, the size of ssnfa1 and asnfa1 was a little higher than PCR product of the nfa1 gene ORF of 360 bp. According to Malhotra et al. (2002), the size of dsRNA and dsDNA was almost double to ssRNA. The reason why size variation was occurred in ssnfa1, asnfa1, and dsnfa1 seemed to be the structural differences of genes. Although the size of synthetic dsnfa1 was not exactly double to single-stranded RNA, because the size of PCR fragment of the nfa1 gene was similar with it of dsRNA, dsnfa1 was successfully synthesized in the present study (Fig. 4A). Moreover, that ssnfa1 and asnfa1 were well synthesized was confirmed by southern blotting of DNA-RNA
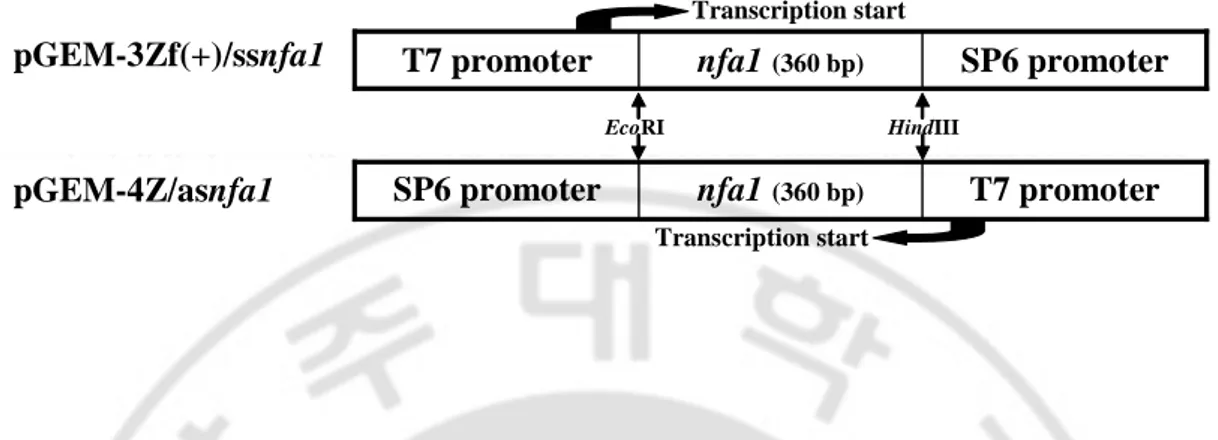
Fig. 3. Schematic representation of an nfa1 gene in N. fowleri used to produce sense (ss) and antisense (as) RNA followed by dsRNA formation in vitro. To
prepare dsRNA, the nfa1 gene cloned into a pGEM–3Zf(+) and pGEM–4Z vector was in vitro transcribed by T7 polymerase, and then ssRNA and asRNA were mixed, incubated at 65℃, and cooled slowly to obtain their corresponding dsRNA.
SP6 promoter nfa1(360 bp) T7 promoter EcoRI HindIII Transcription start pGEM-3Zf(+)/ssnfa1 T7 promoter nfa1 (360 bp) SP6 promoter Transcription start pGEM-4Z/asnfa1
Fig. 4. Findings of dsnfa1 synthesis and Southern blotting. (A) Agarose gel
electrophoresis of the nfa1 gene. To prepare dsnfa1, in vitro transcribed ssnfa1 and
asnfa1 were mixed with equal amounts, incubated at 65℃ and cooled slowly. (B).
Schematic representation of the nfa1 gene of N. fowleri used to produce ssnfa1, asnfa1 and dsnfa1 by in vitro transcription. M, 1kb+ DNA ladder; lane 1, PCR product of nfa1 gene in pGEM–3Zf(+)/ssnfa1; lane 2, PCR product of nfa1 gene in pGEM–4Z/asnfa1; lane 3, nfa1 ssnfa1; lane 4, asnfa1; lane 5, dsnfa1.
A
1.0 0.5 0.3 kbB
1 asnfa1 probe 2 3 1 2 3 ssnfa1 probe M 1 2 3 4 5hybridization (Fig. 4B). DNA of nfa1 gene amplified from pGEM–3Zf(+)/nfa1
vector was detected with α-32P-UTP labeled ssnfa1 or asnfa1. But, a GFP gene in
pEGFP–C2 vector and a control of DW was not detected.
B. Knockdown of the nfa1 mRNA and Nfa1 protein by asnfa1 or dsnfa1
RNAi means post-transcriptional gene silencing and has been induced in most organisms by microinjection. In this present study, a lipid formulation was used for the transfection. Because there have not been reports showing the effect of transfection reagents for RNAi in free-living amoebae, TransMessenger or RNAiFect reagent was used for the transfection of single-stranded and dsRNA. As a northern blot analysis, it was observed that an nfa1 mRNA was successfully knockdowned in
N. fowleri trophozoites transfected with dsnfa1 using RNAiFect reagent (Fig. 5A,
5B). Using TransMessenger reagent, no changes were observed in the level of the
nfa1 gene mRNA. In the principle, TransMessenger reagent is recommended in the
transfection of single-stranded RNA. Hybridizations of northern blotting with a probe specific for the nfa1 gene revealed that in the transfection of asnfa1 using TransMessenger reagent, no changes were observed in the level of the nfa1 gene mRNA and also, in ssnfa1 and only RNAiFect reagent, the level of the nfa1 gene mRNA was almost identical with it of untransfected N. fowleri. Therefore, it was suggested that dsnfa1 should be very effective in knockdowning the nfa1 gene mRNA. An nfa2 gene which used to normalize and show that RNAi was specific for
the nfa1 gene was not changed in the mRNA patterns (Fig. 5A). Therefore, it was
suggested that, although long dsRNA but not siRNA had the problem of non-specificity, dsnfa1 could be specifically acted in N. fowleri. In Fig. 5B, it was shown that there were not any problems in nfa1 gene-specific probe due to the stringent experimental conditions. To more understand the level of knockdowned nfa1 gene mRNA, a quantitative analysis was performed (Fig. 5C). The transfection of dsnfa1 using RNAiFect reagent had the highest effect about 60% on knockdowning the mRNA gene. There were not the differences of dsnfa1 transfected using TransMessenger reagent with asnfa1 transfected using RNAiFect reagent. When asnfa1 was transfected using TransMessenger reagent, no change was also observed in the nfa1 gene mRNA (data not shown).
As a western blot analysis, the Nfa1 protein was not decreased highly as the nfa1 gene mRNA (Fig. 6). It was observed that the Nfa1 protein was higher knockdowned in N. fowleri trophozoites transfected with dsnfa1 than asRNA using RNAiFect or dsnfa1 using TransMessenger reagent (Fig. 6A). Similar amounts of the Nfa1 protein were detected in the controls and the patterns of the Nfa2 protein of 21 kDa were not changed (Fig. 6A). Quantitative estimation of the level of the Nfa1 protein showed higher decreasing effect about 30% in the N. fowleri transfected with dsnfa1 using RNAiFect reagent (Fig. 6B). On contrast, there were few changes in the dsnfa1 using TransMessenger or asnfa1 using RNAiFect reagent. These results indicate that dsnfa1 using RNAiFect reagent is the most efficient for knockdowning the nfa1 and Nfa1 protein
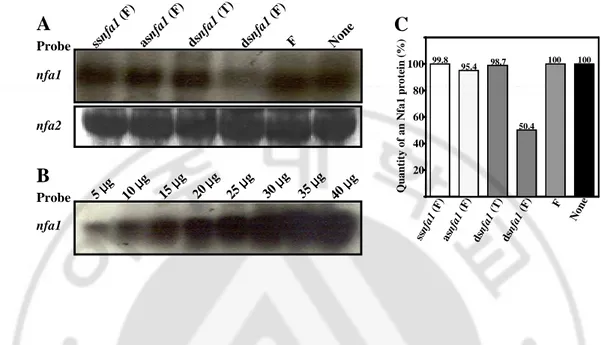
Fig. 5. Northern blotting and quantitative analysis of the nfa1 gene mRNA from
N. fowleri trophozoites transfected with an asnfa1 or dsnfa1. (A) Northern
blotting of the nfa1 or nfa2 gene mRNA with a gene-specific probe. 25 µg of the mRNA samples were loaded. (B) Northern blotting of the nfa1 gene mRNA used as a loading control. C. Quantitative analysis of fig. 5A. F, RNAiFect reagent; T, TransMessenger reagent. Probe nfa1 nfa2 asnf a1 (F ) ssnf a1 (F ) dsnf a1(T ) dsnf a1(F ) F Non e
A
B
5 μμμμg 10 μμμμ g 15 μμμμ g 20 μμμμ g 25 μμμμ g 30 μμμμ g 35 μμμμ g 40 μμμμ gC
Probe nfa1 99.8 95.4 98.7 50.4 100 100 Q u a n ti ty o f a n N fa 1 p ro te in ( % ) 20 40 60 80 100 ssn fa1 (F) asn fa1 (F) dsn fa1 (T) dsn fa1 (F) F Non eFig. 6. Western blotting and quantitative analysis of the Nfa1 protein from N.
fowleri trophozoites transfected with an asnfa1 or dsnfa1. (A) Western blotting of
the Nfa1 or Nfa2 protein detected with a respective polyclonal antibody. 10 µg of the
N. fowleri lysate was loaded. (B) Quantitative analysis of fig. 5A. F, RNAiFect
reagent; T, TransMessenger reagent.
Probe nfa1 nfa2 asnf a1 (F ) ssnf a1 (F ) dsnf a1(T ) dsnf a1(F ) F Non e 99.1 100 97.6 70.1 100 99.8 Q u a n ti ty o f a n N fa 1 p ro te in ( % ) 20 40 60 80 100 ssn fa1 (F) asn fa1 (F) dsn fa1 (T) dsn fa1 (F) F Non e A B
C. Knockdown of an nfa1 gene mRNA and Nfa1 protein by synthetic siRNAs
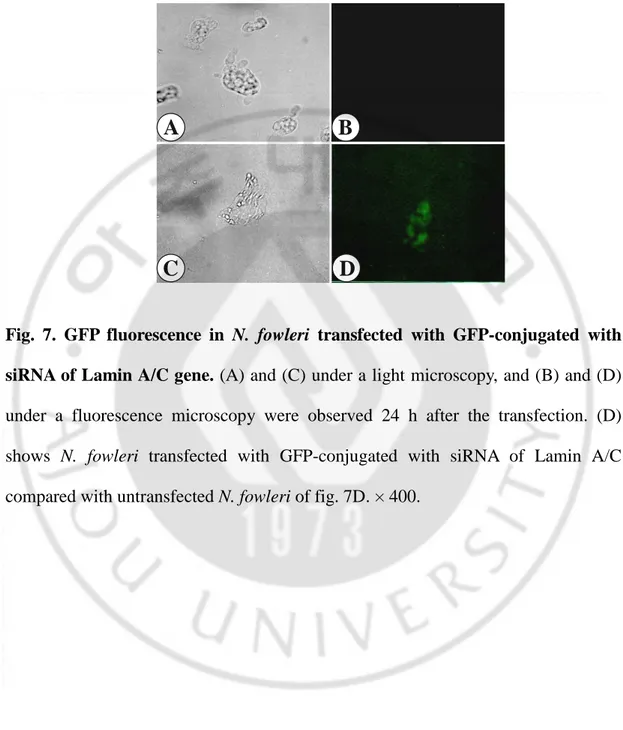
The transfection of siRNA into N. fowleri trophozoites was performed with RNAiFect reagent. It is not easy to find the most functional siRNA of any genes. Although a lot of siRNAs are chosen, they don’t act to knockdown equally. In other words, in the present study, four siRNAs were randomly chosen as considerations of siRNA choices. Because siRNA has very little nucleotide and it is difficult to observe whether the transfection is done or not, we identified the transfection with the fluorescence of GFP-conjugated siRNA of Lamin A/C gene from human, irrespective of the nfa1 gene (Fig. 7). GFP fluorescence was observed 24 h after the transfection into N. fowleri trophozoites with the GFP-conjugated siRNA of Lamin A/C as compared with untransfected N. fowleri. GFP fluorescence was observed in the cytoplasm of transfected N. fowleri. This result showed that siRNA could be transfected using RNAiFect reagent. The fact of the decrease of the nfa1 gene and Nfa1 protein by a dsnfa1 transfection using RNAiFect reagent supported that the RNAi mechanism associated with siRNA not only exist in N. fowleri trophozoites but also siRNA act to knockdown the nfa1 gene. Thus, we transfected each siRNA (sinfa1-1, sinfa1-2, sinfa1-3 and sinfa1-4) using a RNAiFect reagent into N. fowleri trophozoites. All siRNAs were composed of each 21 nucleotides. The expression of the nfa1 gene mRNA in the siRNA transfectants was assessed by northern blot analysis with a specific probe for the nfa1 gene ORF (Fig. 8).
Fig. 7. GFP fluorescence in N. fowleri transfected with GFP-conjugated with siRNA of Lamin A/C gene. (A) and (C) under a light microscopy, and (B) and (D)
under a fluorescence microscopy were observed 24 h after the transfection. (D) shows N. fowleri transfected with GFP-conjugated with siRNA of Lamin A/C compared with untransfected N. fowleri of fig. 7D. × 400.
B
D
A
Although they didn’t show an identical effect, all siRNAs had an effect on knockdowning nfa1 gene mRNA. In particular, the sinfa1-1 showed the highest knockdown of the nfa1 gene mRNA by northern blotting (Fig. 8A). According to the quantitative analysis, even the sinfa1-2 or sinfa1-4 showed the effect from 55 ~ 57% and the sinfa1-1 did the effect about 70% (Fig. 8B). No change of the nfa2 gene mRNA, which is not homologous with the nfa1 gene, was occurred by the transfection of siRNAs of the nfa1 gene (Fig. 8A). Western blots with anti-Nfa1 polyclonal antibody showed the highest decreasing effect of the Nfa1 protein levels by the sinfa1-1, in agreement with the northern blot data (Fig. 9). The Nfa1 protein transfected with each siRNA was not knockdowned as the nfa1 gene mRNA (Fig. 9A). According to the quantitative analysis of fig. 9A, the knockdowned Nfa1 protein was higher about 20% than the nfa1 protein (Fig. 9B). Even though it was done so, the sinfa1-1 showed the highest decreasing effect with about 43%. In the sinfa1-4, the nfa1 gene mRNA was knockdowned with about 45% but the Nfa1 protein with 7%, which was very different from the effect of other three siRNAs. The level of the Nfa2 protein was not affected by the transfection of siRNAs of the nfa1 gene (Fig. 9A). These results supported the functional analysis in vitro. However, when we observed the in vitro cytotoxicity of N. fowleri trophozoites knockdowned by the dsnfa1 or sinfa1-1 against murine macrophages, we didn’t observe the significant inhibition of the cytotoxicity of N. fowleri by the morphological analysis of murine macrophages and LDH release assay (data not shown).
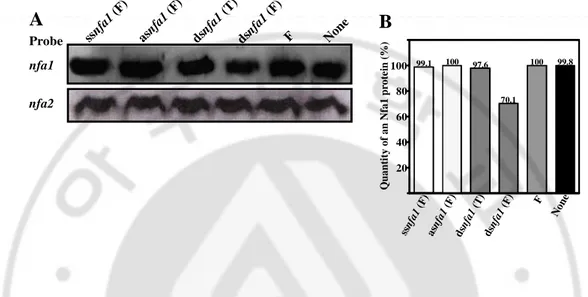
Fig. 8. Northern blotting and quantitative analysis of the nfa1 gene mRNA from
N. fowleri trophozoites transfected with the siRNAs of an nfa1 gene. (A)
Northern blotting of the nfa1 or nfa2 gene mRNA with a gene-specific probe. 25 µg of the mRNA samples were loaded. (B) Quantitative analysis of fig. 5A. N. fowleri trophzoites were transfected using RNAiFect reagent.
sinf a1-1 sinf a1-2 sinf a1-3 sinf a1-4 RN AiF ect Non e Probe nfa1 nfa2 30.8 53.2 44.6 55.4 100 100 Q u a n ti ty o f a n N fa 1 p ro te in ( % ) 20 40 60 80 100 sin fa 1-1 sin fa 1-2 sin fa 1-3 sin fa 1-4 RN AiF ect Non e A B
Fig. 9. Western blotting and quantitative analysis of the Nfa1 protein from N.
fowleri trophozoites transfected with the siRNAs of an nfa1 gene. (A) Western
blotting of the Nfa1 or Nfa2 protein detected with a respective polyclonal antibody. 10 µg of the N. fowleri lysate was loaded. (B) Quantitative analysis of fig. 5A. N.
fowleri trophzoites were transfected using RNAiFect reagent.
sinf a1-1 sinf a1-2 sinf a1-3 sinf a1-4 F Non e Probe nfa1 nfa2 57.5 70.6 66.5 93.6 100 99.3 Q u a n ti ty o f a n N fa 1 p ro te in ( % ) 20 40 60 80 100 sin fa 1-1 sin fa 1-2 sin fa 1-3 sin fa 1-4 RN AiF ect Non e A B