A Case of Von Hippel-Lindau Disease Presented with Multiple Pancreatic Cysts and Medullary Hemangioblastoma
Young Hyun Kim, Hye Lim Jung, Aram Yang, Ji Hee Kwak, Deok Soo Kim, Jung Yeon Shim and Jae Won Shim
Department of Pediatrics, Kangbuk Samsung Hospital, Sungkyunkwan University School of Medicine, Seoul, Korea
Von Hippel-Lindau (VHL) disease is a rare inherited cancer predisposition syndrome characterized by benign and malignant tumors in multiple organs, especially cerebel- lar hemangioblastomas, retinal angiomas, renal-cell carcinoma, and pheochromo- cytomas. Clinically, VHL disease also presents an increased risk for developing multi- ple visceral cysts in the pancreas, liver, and kidneys. Regular surveillance for VHL disease-associated tumors after early diagnosis is necessary for better outcomes in VHL disease. An 11-year-old girl was admitted with prolonged fever lasting for more than 10 days and cervical lymphadenopathy. She did not have a family history of cysts or malignancy. Initial blood tests showed mild leukopenia and moderate ele- vation in aspartate aminotransferase, alanine aminotransferase, and lactate dehydro- genase, but with normal amylase and lipase. Hepatobiliary ultrasonography and mag- netic resonance cholangiopancreatography were done and revealed multiple cysts in- volving the whole pancreas with cyst sizes up to 1.6 cm, indicating VHL disease. Di- rect sequencing of the VHL gene showed a heterozygous duplication at codon 384 (c.384dup), which is predicted to cause a frameshift of the reading frame (p.Leu129Serfs*3).
This was a novel pathogenic variant VHL gene. We carried out the surveillance proto- col for VHL disease-associated tumors, and found a hemangioblastoma in the me- dulla of the brainstem. We are reporting an 11-year-old female patient of VHL dis- ease with brainstem hemangioblastoma who could be suspected and diagnosed of VHL disease in asymptomatic state due to incidentally found multiple pancreatic cysts.
pISSN 2233-5250 / eISSN 2233-4580 https://doi.org/10.15264/cpho.2020.27.1.67 Clin Pediatr Hematol Oncol 2020;27:67∼71
Received on March 30, 2020 Revised on April 21, 2020 Accepted on April 24, 2020
Corresponding Author: Hye Lim Jung Department of Pediatrics, Kangbuk Samsung Hospital, 29 Saemunanro, Jongro-gu, Seoul 03181, Korea Tel: +82-2-2001-2483 Fax: +82-2-2001-1922 E-mail: [email protected] ORCID ID: orcid.org/0000-0003-0601-510X Key Words: Von Hippel-Lindau disease, Pancreatic cyst, Hemangioblastoma
Copyright ⓒ 2020 Korean Society of Pediatric Hematology-Oncology
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/
Introduction
Von Hippel-Lindau (VHL) disease is a rare autosomal dominantly inherited familial cancer-predisposition syn- drome with an incidence of 1 in 36,000 live births. VHL disease is caused by a germline mutation of VHL tu- mor-suppression gene located on chromosome 3p25-26 [1]. The VHL gene mutation predisposes patients to de-
velopment of multiple cysts and tumors in various organs [2]. The most commonly reported cysts and tumors in VHL disease are cerebellar and spinal hemangioblastomas (HBs, 60-80%), retinal angiomas or capillary HBs, pan- creatic neuroendocrine tumors (PNETs, 20%), pancreatic cysts (75%), clear-cell renal-cell carcinoma (RCC, 24- 45%), renal cysts (38%), pheochromocytoma (20%), and endolymphatic sac tumors (ELSTs, 10-15%) [2-5]. Cerebellar HB usually presents at early adulthood and causes in-
creased intracranial pressure, and spinal cord HB causes proprioception abnormalities and disturbances of gait and bladder function. Approximately 25% of VHL disease with cerebellar HBs also have retinal angiomas, which can cause retinal detachments and loss of visual acuity.
Although central nervous system (CNS) and retinal HBs contribute to morbidity, RCCs are the most common cause of death, causing 50% of death in VHL disease [5].
Although 80% of VHL disease are autosomal domi- nantly inherited from an affected parent, 20% of VHL dis- ease cases are a de novo gene mutation without family history [6]. VHL disease-associated tumors usually pres- ent clinically in early adulthood, third to fourth decade, and more than 90% of penetrance is presented by the age of 65 [7,8]. Early diagnosis of VHL disease and regular follow-up with appropriate evaluations according to sur- veillance guidelines for VHL disease are necessary to identify pathologic lesions that may be treated at an ear- ly stage [5].
Herein, we are reporting a genetically confirmed case of VHL disease with brainstem HB in an 11-year-old girl without a family history who could be suspected and di- agnosed of VHL disease in asymptomatic state due to in- cidentally found multiple pancreatic cysts.
Case Report
An 11-year-old girl was admitted with prolonged fever lasting for more than 10 days, sore throat, and cervical lymphadenopathy. She did not have a past medical his- tory of admission or family history of cysts or malign- ancy.
On admission, she had a fever (temperature up to 38.5oC), and physical examination showed throat injection with palatine tonsillar hypertrophy, multiple cervical lymph-node enlargement on both neck levels II and III with a size smaller than 1.5 cm. Other physical examina- tion findings including a neurologic examination were normal.
Initial CBC showed WBC 3,750/µL, Hb 13.7 g/dL, pla- telet 293,000/µL, absolute neutrophil count 2,088/µL, re- vealing mild leukopenia. Blood chemistry showed ele-
vation in liver enzymes, including aspartate amino- transferase (AST, 111 IU/L), alanine aminotransferase (ALT, 208 IU/L), and lactate dehydrogenase (LDH, 412 IU/L), but amylase (75 U/L) and lipase (16 U/L) were normal. For the work-up of fever and hepatitis, C-re- active protein and cultures in blood, urine, cerebrospinal fluid, and throat were all negative. Virus studies and im- munologic studies yielded negative results.
Neck ultrasonography (USG) was done for cervical lymph- node enlargement, and she was diagnosed as having be- nign reactive cervical lymphadenitis. Hepatobiliary USG was done, and multiple pancreatic cysts and mild fatty liver were observed (Fig. 1). For further evaluation of the pancreatic cysts, we did magnetic resonance cholangio- pancreatography (MRCP), which showed multiple cystic lesions involving the whole pancreas, with cyst sizes up to 1.6 cm, but no neuroendocrine tumors of the pan- creas, other tumors, or renal cysts were observed (Fig. 2).
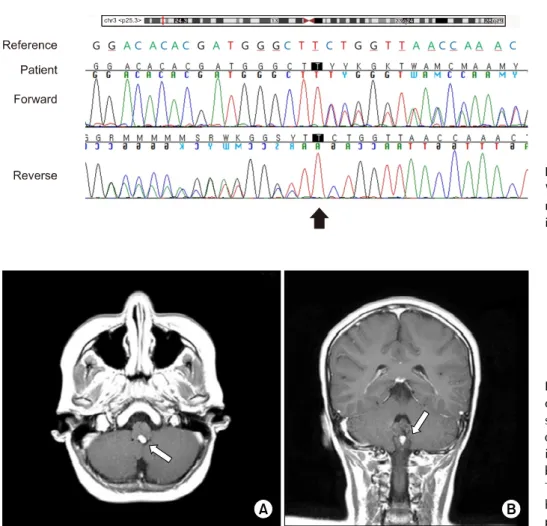
The MRCP finding implied an association of a genetic disorder, such as VHL disease. Therefore, VHL tumor- suppressor gene direct sequencing analysis and related tumor markers, including serum carcinoembryonic anti- gen (CEA) and carbohydrate antigen 19-9 (CA 19-9) stud- ies were performed. Her CEA and CA 19-9 level were normal. Sequence analysis for the VHL gene showed het- erozygous for duplication at codon 384 (c.384dup) in exon 2 of the VHL gene on chromosome 3p25.3, which is predicted to cause a frameshift of the reading frame (p.Leu129Serfs*3) (Fig. 3). This variant was assessed as a novel pathogenic variant based on the 2015 American College of Medical Genetics and Genomics and the Association for Molecular Pathology guidelines: PVS1 (a frameshift variant that leads to a truncated protein) and PM2 (absent from controls in the Exome Aggregation Consortium and Genome Aggregation Database), and PP4 (highly specific patient phenotype) [9].
The fever subsided after 16 days, and a follow-up lab- oratory examination showed improvement in leukope- nia, AST, ALT, and LDH level before discharge on the 9th day of hospitalization, which was finally normalized 4 months later at our outpatient clinic.
After genetic confirmation as VHL disease, we did sur-
Fig. 1. Hepatobiliary ultrasonogra- phy showing (A) mild fatty liver with possibly focal fat sparing zones and (B-D) a few anechoic lesions in pancreas, size up to 1.4 cm, im- plicating multiple pancreatic cysts (arrows).
Fig. 2. Magnetic resonance cholangiopancreatography (MRCP) showing multiple cystic lesions involving whole pancreas, with cyst size up to 1.6 cm, without definite evidence of mural no- dule or direct communication of the main pancreatic duct (ar- row). This MRCP finding of multiple pancreatic cysts implicate the association of genetic disorder such as von Hippel-Lindau dis- ease in this patient. There were no anatomical variation of bili- ary tree or cysts in both kidneys.
veillance for VHL disease-associated tumors. In brain magnetic resonance imaging (MRI), a 9.2×4.5×8.1 mm sized well-defined enhancing lesion was detected in the posterior aspect of the medulla of the brainstem, sus- pected of HB (Fig. 4). After consultation with neuro- surgery, we planned to follow up with a brain MRI every
year and considered surgical excision if symptoms devel- oped or size of the HB increases.
Discussion
The VHL gene is a tumor-suppressor gene located on chromosome 3p25-26, which is composed of 3 exons en- coding 2 isoforms of protein, pVHL [5,10].
VHL disease is classified as type 1 and type 2 with dif- ferent genotype-phenotype manifestation. Type 1 VHL results from a loss-of-function mutation, such as trunca- tion mutation of the VHL gene, and is characterized by rare development of pheochromocytoma. Type 2 VHL results from a missense mutation of the VHL gene and is characterized by development of pheochromcytoma.
Type 2 VHL is further subdivided into type 2A VHL with- out RCC development, type 2B VHL with RCC develop- ment, and type 2C VHL with only pheochromcytoma development. From the VHL gene mutation type, our case with a truncation mutation of the VHL gene can be classified as type 1 VHL [11].
The mean age of initial tumor diagnosis in VHL disease is 26 years (range 1-70 years) [7], but different tumors develop typically at different mean ages. Cerebellar HBs are diagnosed in the 3rd decade (at a mean age of 29 years), and brainstem and spinal HBs develop rarely in
Fig. 3. Sequence analysis of the VHL gene. A heterozygous novel va- riant of c.384dup (p.Leu129Serfs*3) in exon 2 is noted.
Fig. 4. (A) Axial and (B) coronal view of brain magnetic resonance imaging showing 9.2x4.5x8.1 mm sized well defined enhancing lesion in poster- ior aspect of medulla, most pro- bably hemangioblastoma (arrows).
There are no evidence of stroke, hemorrhage, atrophy or hydroce- phalus in the brain.
the younger population (less than 40 years of age), but in multiple location. RCCs are diagnosed in the 4th dec- ade, but can be as early as at 16 years of age, presenting with hematuria, flank pain, or a palpable mass. Pheo- chromocytomas are diagnosed at a mean age of 28 years, presenting with secondary hypertension or stroke. Pan- creatic lesions in VHL disease are diverse, including sim- ple cysts within pancreatic parenchyma, serous cys- tadenomas, and PNETs. Simple pancreatic cysts are be- nign and typically asymptomatic, but surgical treatment is indicated when they are symptomatic because of com- pression or obstruction.
Surveillance guidelines have been developed by the VHL disease Family Alliance and are recommended for VHL disease patients diagnosed by VHL gene analysis and for patients’ family members who have not undergone VHL gene analysis [5]. In individuals diagnosed with the VHL gene mutation, there should be a yearly assessment
of neurologic status, visual acuity and ophthalmologic status, hearing, and blood pressure. After the age of 5 years, there should be laboratory screening for pheo- chromocytoma every year, hearing evaluation every 2 years, and contrast-enhanced MRI for CNS and spine ev- ery 2 years [11,12]. Contrast-enhanced MRI for CNS with thin cuts of the internal auditory canal is necessary to evaluate the ELST for symptomatic patients [11,12]. After the age of 16 years, there should be abdominal USG yearly to identify visceral lesions, and MRI of the abdo- men and entire neural axis every 2 years [11,12].
Our patient could be suspected and genetically diag- nosed as VHL disease because of incidentally found mul- tiple pancreatic cysts by abdominal USG and MRCP. By using the surveillance protocol for VHL disease-associ- ated tumors, we could diagnose brainstem HB before progression. We plan to conduct a VHL gene analysis and surveillance protocol for VHL disease-associated tumors
in other family members of the patient and carry out reg- ular follow-up surveillance in our patient.
In one previous study of 55 VHL disease in Korea, there were 7 pediatric cases genetically diagnosed at age of 10 to 18 years [13]. Amongst 7 pediatric patients, 4 patients manifested as CNS HB and either pancreatic cysts or tumor, or renal or hepatic cyst, and 3 patients manifested as either retinal or CNS HB [13]. In another study of 26 VHL disease in Korea, there were 3 pediatric cases genetically diagnosed at age of 12, 11 and 14 years [14]. Amongst 3 pediatric patients, 12 year old male pa- tient with family history manifested with CNS HB, RCC, pancreatic lesion and renal cyst, 11 year old male patient manifested with symptomatic CNS HB, pancreatic and renal cysts, and 14 year old male patient manifested with symptomatic pancreatic cysts and retinal HB [14]. In our 11-year-old VHL disease case without family history, multiple pancreatic cysts found incidentally in asympto- matic state became a clue to VHL disease. When there is symptomatic or asymptomatic multiple pancreatic cysts with or without renal or hepatic cysts, VHL disease must be suspected and surveillance protocol for VHL dis- ease-associated tumors is recommended.
Conflict of Interest Statement
The authors have no conflict of interest to declare.
References
1. Keutgen XM, Hammel P, Choyke PL, Libutti SK, Jonasch E, Kebebew E. Evaluation and management of pancreatic le- sions in patients with von Hippel–Lindau disease. Nat Rev
Clin Oncol 2016;13:537-49.
2. Sharma A, Mukewar S, Vege SS. Clinical profile of pancreatic cystic lesions in von Hippel-Lindau disease: A series of 48 patients seen at a tertiary institution. Pancreas 2017;46:948- 52.
3. Bhuyan M, Dutta D, Baishya BK, Hussain Z. Cerebellospinal hemangioblastoma with bilateral pheochromocytoma and hepatic cyst: a rare entity. Asian J Neurosurg 2016;11:311.
4. Ayloo S, Molinari M. Pancreatic manifestations in von Hippel– Lindau disease: a case report. Int J Surg Case Rep 2016;21:
70-2.
5. Findeis-Hosey JJ, McMahon KQ, Findeis SK. Von Hippel-Lindau disease. J Pediatr Genet 2016;5:116-23.
6. Richards FM, Payne SJ, Zbar B, Affara NA, Ferguson-Smith MA, Maher ER. Molecular analysis of de novo germline muta- tions in the von Hippel-Lindau disease gene. Hum Mol Genet 1995;4:2139-43.
7. Lonser RR, Glenn GM, Walther M, et al. von Hippel-Lindau disease. Lancet 2003;361:2059-67.
8. Wilding A, Ingham SL, Lalloo F, et al. Life expectancy in he- reditary cancer predisposing diseases: an observational study.
J Med Genet 2012;49:264-9.
9. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus re- commendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
Genet Med 2015;17:405-24.
10. van Asselt SJ, de Vries EG, van Dullemen HM, et al. Pancreatic cyst development: insights from von Hippel-Lindau disease.
Cilia 2013;2:3.
11. Rednam SP, Erez A, Druker H, et al. Von Hippel-Lindau and hereditary pheochromocytoma/paraganglioma syndromes:
clinical features, genetics, and surveillance recommendations in childhood. Clin Cancer Res 2017;23:e68-e75.
12. Ganeshan D, Menias CO, Pickhardt PJ, et al. Tumors in von Hippel-Lindau syndrome: from head to toe-comprehensive state-of-the-art review. Radiographics 2018;38:849-66.
13. Hwang S, Ku CR, Lee JI, et al. Germline mutation of Glu70Lys is highly frequent in Korean patients with von Hippel-Lindau (VHL) disease. J Hum Genet 2014;59:488–93.
14. Lee JS, Lee JH, Lee KE, et al. Genotype-phenotype analysis of von Hippel-Lindau syndrome in Korean families: HIF-α binding site missense mutations elevate age-specific risk for CNS hemangioblastoma. BMC Med Genet 2016;17:48.