DOI 10.17480/psk.2018.62.5.297
Amelioration of DSS-induced colitis by BJ-2223 through the inhibition of Th17 cell differentiation
Maheshwor Timilshina and Jae-Hoon Chang
#College of Pharmacy, Yeungnam University, Gyeongsan 38541
(Received September 5, 2018; Revised September 13, 2018; Accepted September 13, 2018)
Abstract — CD4
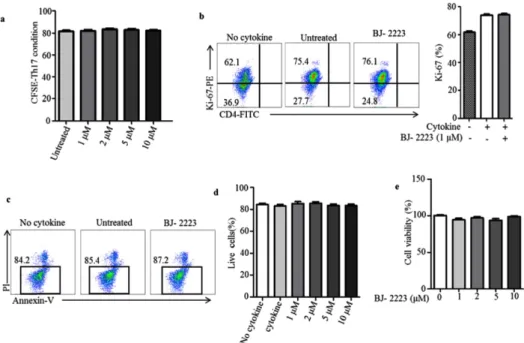
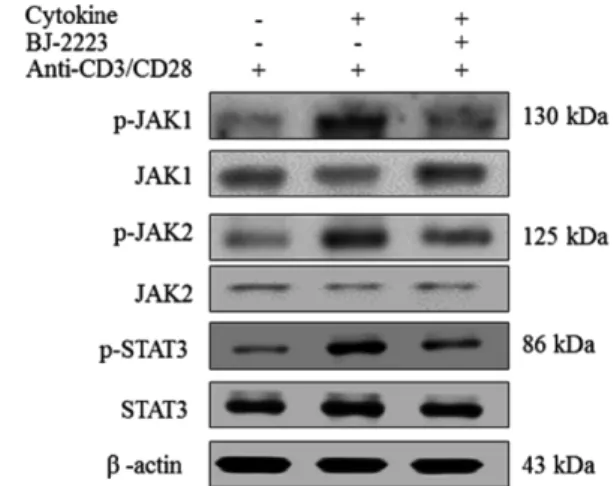
+T cells differentiate into T helper (Th) 1 and Th17 cells, which are key mediators of several inflam- matory and autoimmune diseases. Inhibition of Th1 and Th17 cell differentiation helps attenuate several inflammatory con- ditions. Th17 cells are highly pro-inflammatory cells that orchestrate tissue inflammation and ulcerative colitis (UC). Thus, novel compounds that inhibit Th17 cell differentiation are required for the effective treatment of inflammatory and auto- immune diseases. By extensive screening of several synthetic compounds, we found that BJ-2223 inhibits Th17 cell dif- ferentiation without affecting Th1 and regulatory T (Treg) cell differentiation in vitro. Furthermore, BJ-2223 does not induce apoptosis and does not affect T cell proliferation and viability. Mechanistically, BJ-2223 inhibits Th17 cell differentiation through inhibition of the Janus-activated kinase (JAK)/signal transducer and activator of transcription protein (STAT) sig- naling pathway. Moreover, BJ-2223 ameliorates dextran sulfate sodium (DSS)-induced colitis, protects intestinal tissue, and decreases the percentage of Th17 cells in vivo. Thus, BJ-2223 is a novel compound that inhibits in vitro Th17 differentiation and attenuates DSS-induced colitis by reducing the percentage of Th17 cells.
Keywords BJ-2223, Th17 cell, Differentiation, JAK/STAT, DSS-induced colitis
Introduction
Upon encountering an antigen/MHC complex, CD4
+T cells are activated and polarized to Th1, Th17, and Treg cells, depending on the cytokine milieu of the microenvironment.
1)Th1 and Th17 cells are involved in eliciting immune responses against extracellular pathogens and bacterial and fungal infec- tion.
2,3)T cell receptor (TCR) activation in the presence of specific cytokine guidance, induces the proliferation and differ- entiation of naïve CD4
+T cells into several lineages of Th cells that can be distinguished by their unique cytokine profile and functions.
4)The binding of interferon-(IFN)-γ and interleu- kin (IL)-12 activates signal transducer and activator of tran- scription protein (STAT)1 and STAT4, thereby activating Th1- specific T-box transcription factor (T-bet) and drives the differ- entiation of naïve CD4
+T cells into Th1 cells.
5)Differentia- tion of Th17 cells is driven by STAT3-activating cytokines,
such as IL-6, IL-21, and IL-23, which upregulate the expres- sion of the transcription factor retinoid-related orphan recep- tor (ROR) γt.
6)IL-2-mediated STAT5 activation leads to the differentiation of naïve CD4
+T cells to Foxp3
+Treg cells, which are necessary for the prevention of autoimmune dis- eases and maintenance of immune homeostasis.
7)However, the abnormal activation and differentiation of Th1 and Th17 cells lead to different autoimmune inflammatory diseases.
Inflammatory bowel diseases (IBDs), characterized by ulcer- ative colitis (UC) and Crohn’s disease (CD), are progressive and chronic inflammatory disorders that are caused due to an imbalanced mucosal immune system.
8)IBD pathogenesis is associated with the increase in the concentration of inflamma- tory mediators and cytokines, such as IFN- γ and IL-6.
9,10)IL-6 is the key cytokine that promotes the generation and differen- tiation of Th17 cells.
11)Moreover, IL-6 intensifies IBD by enhancing T cell survival and apoptosis resistance at the site of inflammation.
12)Th17 cells are widely present throughout the intestinal lamina propria and provide protection against the pathogens in the mucosal surrounding, but sustained activa- tion of Th17 cells causes colitis.
13)Dextran sulfate sodium (DSS) is the most widely studied mouse model of IBD because
#