저작자표시-비영리-변경금지 2.0 대한민국 이용자는 아래의 조건을 따르는 경우에 한하여 자유롭게 l 이 저작물을 복제, 배포, 전송, 전시, 공연 및 방송할 수 있습니다. 다음과 같은 조건을 따라야 합니다: l 귀하는, 이 저작물의 재이용이나 배포의 경우, 이 저작물에 적용된 이용허락조건 을 명확하게 나타내어야 합니다. l 저작권자로부터 별도의 허가를 받으면 이러한 조건들은 적용되지 않습니다. 저작권법에 따른 이용자의 권리는 위의 내용에 의하여 영향을 받지 않습니다. 이것은 이용허락규약(Legal Code)을 이해하기 쉽게 요약한 것입니다. Disclaimer 저작자표시. 귀하는 원저작자를 표시하여야 합니다. 비영리. 귀하는 이 저작물을 영리 목적으로 이용할 수 없습니다. 변경금지. 귀하는 이 저작물을 개작, 변형 또는 가공할 수 없습니다.
Studies on the Mechanism of
Polyamine and Prothrombin
kringle-2-induced Degeneration of
Neuron and Microglia
by
So Yoon Won
Major in Neuroscience
Department of Medical Science
The Graduate School, Ajou University
Studies on the Mechanism of
Polyamine and Prothrombin
kringle-2-induced Degeneration of
Neuron and Microglia
by
So Yoon Won
A Dissertation Submitted to The Graduate School of
Ajou University in Partial Fulfillment of the
Requirements for the Degree of
Ph. D. in Neuroscience
Supervised by
Byung Kwan Jin, Ph.D.
Eun Joo Baik , MD Ph.D.
Major in Neuroscience
Department of Medical Science
The Graduate School, Ajou University
This certifies that the dissertation
of So Yoon Won is approved.
SUPERVISORY COMMITTEE
Eun Hye Joe
.
Yong Beom Lee
.
Byung Gon Kim
.
Eun Joo Baik
.
Byung Kwan Jin
.
The Graduate School, Ajou University
June, 25th, 2010
i
ACKNOWLEDGEMENT
(감사의 글)
인생의 터닝포인트에서 삶을 돌아보니 제게 있어 학위과정의 길은 학
문의 길보다는 어쩌면 인격수양의 과정에 더 가깝지 않았나 싶습니다.
이제 비로소 모든 과정을 마치고 논문의 마지막 마무리를 글로 남기
려 하니 옛일이 스쳐 지나가면서 베풀지는 못하고 받기만 한 삶을 반
성하게 됩니다. 저를 도와 주신 분이 이렇게도 많았음에도 불구하고
일일이 찾아 뵙고 감사 드리지 못하는 점 용서를 구합니다. 참으로 부
족한 저를 학문의 길로 불러 들여 오늘의 제가 있게 해주신 평생 스
승인 지도교수 진병관 교수님의 은혜에 고개 숙여 깊이 감사 드립니
다.
같은 지도교수님 지도하에 졸업하신 최상호 선배, 이다용 선배, 김상
룡 선배 ,정은숙 선배, 박근우 선배님 들에게도 감사의 말을 전합니다.
어렵고 힘들었던 시절 동고동락한 실험실동기 영철오빠의 박사 학위
취득을 진심으로 축하합니다. 우리 실험실 식구들 역시 제게 큰 도움
을 주었습니다. 실험실 각종 자질구레한 일들을 군소리 하나 없이 처
리하는 은수오빠, 아이낳고 몸조리 하는 수희, 똑 소리 나는 유진이,
오랜 시간 같이 하지 못했지만 잠시 동안 짝궁 으로 지낸 막내 진한
이 모두에게 감사의 말을 전하고 싶습니다. 모두들 앞날에 무궁한 영
광이 깃들기를 기원합니다.
제가 어린 시절부터 늘 사랑과 관심으로 내 인생의 든든한 후원자가
되어주신 엄마, 아빠의 희생과 사랑이 아니였으면 제가 오늘 이 자리
에 없었을지도 모릅니다. 부모님께는 죄송하다는 말씀과 감사하다는
말씀을 동시에 드리려 합니다.
두 아들의 육아를 부탁드려 늘 고단하게 해드린 점 송구스럽게 생각
하며, 또 사랑으로 아이들을 돌보아 주셔서 아이들이 밝게 자라게 해
주신 점 다시 한번 감사 드리고 사랑합니다. 내 사랑하는 동생 정용이
에게도 감사 드립니다.
ii
공부한다는 핑계로 집안일에는 하나도 신경 못씀에도 아무 말씀 하
지 않으시고 늘 그 자리에서 힘이 되어주시고, 무슨 일이든 믿어주고,
격려해 주고 아낌없이 지원을 보내준 시부모님 에게 뭐라 감사의 말
을 할지 모르겠습니다. 감사하고 사랑합니다.
그리고 어려운 일이나 슬픈일 도 언제나 함께 하는 사랑하는 남편, 언
제나 내꿈의 언저리에서 맴도는 눈에 넣어도 아프지 않을, 사랑스런
내 인생의 보배 진우와 진영이. 진우야 진영아 어려운 일 앞에서도 당
당하게 헤쳐 나가는 사람이 되기를 바란다. 항상 함께한 가족이 있어
서 든든했고, 앞으로 더 좋은 일들, 더 행복한 일들이 많아지기를 바
랍니다.
일일이 거론하지는 못했지만 저를 아는 모든 분들께 감사드리며, 이
논문을 바칩니다.
iii
-ABSTRACT-
Studies on the Mechanism of Polyamine-and Prothrombin
kringle-2-induced Degeneration of Neuron and Microglia
Neural cell death is the defining feature of all neurodegenerative diseases and is the underlying cause of many functional deficits. Clarifying the mechanism of cell death in the nervous system is essential to understand disease pathology and to devise effective treatment strategies. Increasing evidence has linked chronic inflammation to a number of neurodegenerative disorders including Alzheimer’s disease (AD), Parkinson’s disease (PD). In the central nervous system, microglia, the resident innate immune cells play major role in inflammatory process. Microglia may directly toxic to neurons by releasing various substances such as inflammatory cytokines. Thus, it is pathophysiologically important to understand how the extent and duration of brain inflammation is controlled in vivo. In addition to down-regulation of inflammatory mediators, the extent and duration of inflammation in the CNS may be controlled by the removal of activated microglia. Therefore, this study shows the degeneration mechanism of dopaminergic neuron and the rolls of activated microglia to provocation or prevention of neuronal cell death.
Recently polyamines have been also implicated in cell death. I examined whether polyamines, endogenous ligand for transient receptor potential vanilloid 1 (TRPV1), could mediate 6-hydroxydopamine (6-OHDA)-induced Mesencephalic Dopaminergic
iv
(DA) Neurons In vivo and In Vitro. Intranigral injection of the 6-OHDA into the rat brain, or treatment of rat mesencephalic cultures with 6-OHDA, resulted in cell death of dopaminergic (DA) neurons, as visualized by immunocytochemistry. This in vivo and in vitro effect was prevented by the α-difluoromethylornithine (DFMO), an inhibitor of polyamines biosynthesizing enzyme ornithine decarboxylase (ODC), suggesting the direct involvement of Polyamines in neurotoxicity. TRPV1 antagonist capsazepine (CZP) reversed 6-OHDA-or polyamines-induced loss of DA neurons in vivo and in vitro, indicating TRPV1-mediated neurotoxicity. Western blot analysis also showed that pretreatment with DFMO reversed 6-OHDA-induced downregulation of phosphorylated Akt and upregulation of cleaved caspase-3 in mesencephalic cultures. This result is the first to show that 6-OHDA-polyamines-TRPV1 signaling pathway exerts neurotoxicity on dopaminergic neurons in vivo and in vitro. Prothrombin kringle-2 acts as an proinflammtory agent that activates microglia to release pro-inflammatory mediators. Intracortical injection of pKr-2 caused significant loss of cortical neurons in vivo after seven days, as evident from Nissl staining and immunohistochemical analysis using the neuronal-specific nuclear protein (NeuN) antibody. In parallel, pKr-2-activated microglia and ROS production were observed in rat cortex displaying degeneration of cortical neurons. Reverse transcription-PCR at various time points after pKr-2 administration disclosed early and transient expression of inducible nitric oxide synthase (iNOS) and proinflammatory cytokines, such as interleukin 1β (IL-1β). Co-localization of iNOS, IL-1β, and TNF-α within microglia was evident with double-label
v
immunohistochemistry. Additionally, pKr-2 induced upregulation of cytosolic components of NADPH oxidase (p67phox), translocation of cytosolic p67phox protein to the membrane, and p67phox expression in microglia in the cortex in vivo, signifying NADPH oxidase activation. The pKr-2-induced oxidation of proteins and loss of cortical neurons were partially inhibited by DPI, an NADPH oxidase inhibitor, and trolox, an antioxidant. Consistent with my hypothesis, following treatment with pKr-2 in vitro, neurotoxicity was detected exclusively in co-cultures of cortical neurons and microglia, but not in microglia-free neuron-enriched cortical cultures, indicating that microglia are required for pKr-2 neurotoxicity. My results strongly suggest that pKr-2 as an endogenous compound participates in cortical neuron death through microglial NADPH oxidase-mediated oxidative stress. Therefore, inhibition of microglial NADPH oxidase activation may offer prospective clinical therapeutic benefit for neuroinflammation-related neurodegenerative disorders. How to minimize brain inflammation is pathophysiologically important, since inflammation induced by microglial activation can exacerbate brain damage. Thus, Death of activated microglia could act as a critical mechanism for the resolution of brain inflammation. Microglia cell death was detected at eight days after co-treatment of pKr-2 with IL-13/IL-4 in vitro. This cell death assessed by live and dead assay, TUNEL and MTT assay. Interestingly, superoxide assay, WST-1 show significantly increased reactive oxygen species (ROS) in combination of pKr-2 and IL-13 or IL-4 treated microglia. Additionally, the IL-13/IL-4-enhanced ROS were partially inhibited by an NADPH
vi
oxidase inhibitor. The IL-13/IL-4 induced cell death of microglia were partially inhibited by an NADPH oxidase inhibitor and by an antioxidant. Therefore, I hypothesized that the effects of IL-13/IL-4 and NADPH oxidase-derived ROS production may be linked to death of activated microglia. Moreover, Western blot analysis show significantly increased Cyclooxygenase-2 (COX-2) expression in combination of pKr-2 and IL-13 or IL-4 treated microglia. The IL-13/IL-4-enhanced COX-2 expression were partially inhibited by an NADPH oxidase inhibitor and an antioxidant. Additional studies demonstrated that microglia cell death was reversed by treatment with NADPH oxidase inhibitor, antioxidant, and COX-2 inhibitor. To my knowledge, This result is the first to demonstrate that IL-13/IL-4 induced cell death of pKr-2 activated microglia is mediated by oxidative stress and COX-2 through NADPH oxidase.
Key words: Dopamiergic neuron, Polyamines, 6-hydroxydopamine (6-OHDA), Transient receptor potential vanilloid subtype 1 (TRPV1), Ornithine decarboxylase (ODC), α-difluoromethylornithine (DFMO), Prothrombin kringle-2 (pKr-2), Microglia, Interlekin-13 (IL-13), Interleukin-4 (IL-4)
vii
TABLE OF CONTENTS
ACKNOWLEDGEMENT ··· i
ABSTRACT ··· iii
TABLE OF CONTENETS ··· vii
LIST OF FIGURES ··· x
ABBREVIATION ··· xii
I. INTRODUCTION ··· 1
II. MATERIALS AND METHODS A. METHODS 1. Sterotaxic surgery and drug injection ··· 16
2. Tissue preparation and immunohistochemistry ··· 16
3. Double-immunofluorescence staining ··· 18
4. Live and dead assay ··· 19
5. MTT Reduction Assay ··· 19
6. TdT-mediated dUTP Nick-EnD Labeling (TUNEL) assay ··· 20
7. Immunocytochemistry ··· 20
8. Reverse transcription-polymerase chain reaction (RT-PCR) ··· 21
9. Western blot analysis ··· 21
10. In situ detection of O2- and O2- 11. Detection of protein oxidation ··· 24
viii
12. Measurement of ROS Generation ··· 25
13. Primary culture of cortical neuron, drug treatement, and assessment neuronal cell death ··· 25
14. Primary culture of dopaminergic neuron, drug treatement, and assessment neuronal cell death ··· 26
15. Cortical Microglia Cultures ··· 27
16. Co-cultures of microglia and neurons ··· 27
17. ODC enzyme activity assay ··· 28
18. Dopamine (DA) uptake assay ··· 28
19. Measurement of the densities of the immunoblot bands ··· 29
20. Determination of NO ··· 29
21. Quantification of TNF-α Release ··· 29
22. Statistical analysis ··· 29
III. RESULTS PART I. DFMO, Ornithine Decarboxylase Inhibitor, Prevents 6- hydroxydopamine-Induced Neurotoxicity in Rat Mesencephalic Dopaminergic Neuron 1. Polyamine depletion protects on 6-OHDA-induced neurotoxicity of dopainergic neurons in neuron-enriched mesencephalic culture ··· 30
2. ODC activity in neuron-enriched mesencephalic culture exposed to 6-hydroxydopamine (6-OHDA) ··· 34
ix
3. Spermidine and Spermine induces neurotoxicity of dopaminergic neurons in neuron-enriched mesencephalic culture ··· 36 4. Polyamines and 6-OHDA induces loss of dopaminergic neurons through
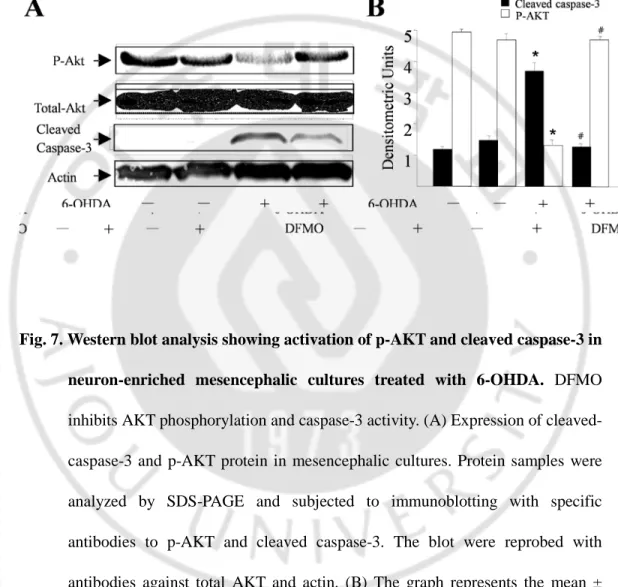
TRPV1 in neuron-enriched mesencephalic culture ··· 38 5. Effects of DFMO on the phosphorylation of AKT and activation of
caspase-3 in neuron-enriched mesencephalic culture exposed to 6-OHDA · 40 6. Depletion of Polyamines rescue dopaminergic neurons in the SN from
6-OHDA-induced neurotoxicity in vivo ··· 42
PART II. Prothrombin kringle-2-induced oxidative stress contributes to the death of cortical neurons in vivo and in vitro:Role of microglial NADPH oxidase.
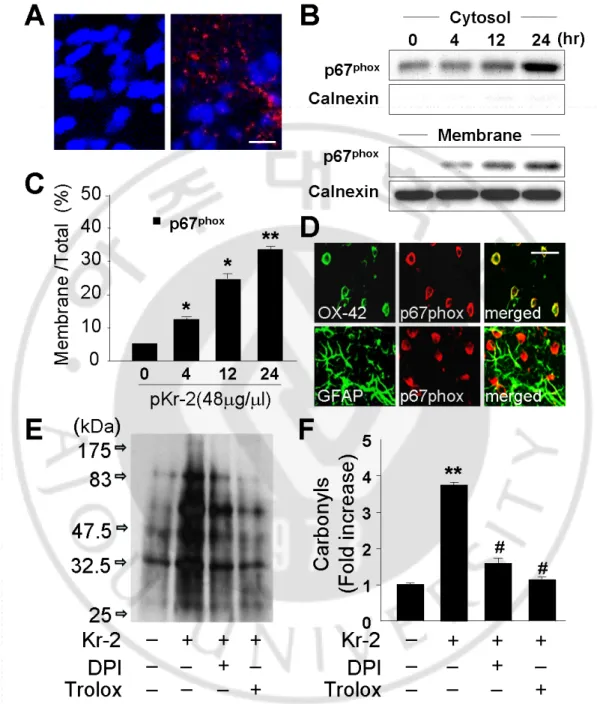
1. pKr-2 induces microglial activation and proinflammatory cytokines in the cortex in vivo ··· 45 2. pKr-2 stimulates NADPH oxidase-mediated production of ROS by
activated microglia in the cortex ··· 51 3. NADPH oxidase contributes to pKr-2-induced neurodegeneration in the
cortex in vivo ··· 56 4. pKr-2 induces cortical neuron death in co-cultures of neuron and microglia .58
x
PART III. IL-13/IL-4 regulate oxidative stress via activation of NADPH oxidase and cell death of microglia in Prothrombin kringle-2-treated microglia culture
1. IL-13/IL-4 induced cell death of cultured rat microglia ··· 61
2. Interleukin-13/-4 enhances the NADPH Oxidase-Mediated Production of ROS in pKr-2 treated microglia cultures ··· 64
3. NADPH oxidase contributes to IL-13/IL-4-induced death of activated microglia In vitro ··· 66
4. Interleukin-13/-4-enhanced COX-2 contributes to microglia cell death mediated by the NADPH oxidase-mediated production of ROS ··· 69
IV.DISCUSSION ··· 72
V.CONCLUSION ··· 87
REFERENCES ··· 88
xi
LIST OF FIGURES
- PART I-Fig.1. Structure of polyamines ··· 4 Fig.2. Relative activation and permeation of TRPV1 by polyamines ··· 10 Fig.3. Effects of DFMO, ornithine decarboxylase on 6-hyroxydopmaine
(6-HDA)-induced neurotoxicity of dopaminergic neurons in neuron-enriched mesencephalic culture ··· 32 Fig.4. ODC activity in dopaminergic neurons exposed to 6-hyroxydopmaine (6-OHDA) ··· 35 Fig.5. Spermidine (SPD) and Spermine (SPM) induces cell death of dopaminergic
neurons in mesencephalic cultures ··· 37 Fig.6. Effects of CZP (TRPV1 antagonist) on 6-OHDA (30 μM) or Spermidine
(SPD) and Spermine (SPM)-induced neurotoxicity in dopaminergic neurons in neuron-enriched mesencephalic culture ··· 39 Fig. 7. Western blot analysis showing activation of p-AKT and cleaved caspase-3
in neuron-enriched mesencephalic cultures treated with 6-OHDA ··· 41 Fig.8. TH-immunohistochemistry showing protective effects of DFMO on the
6-OHDA- induced degeneration of dopaminergic neurons in the SN ··· 43
- PART II-
xii
Fig.10. pKr-2-induced iNOS, IL-1β, and COX-2 protein expression in activated microglia ··· 49 Fig.11. p-2 induces O2
-Fig.12.Oxidative stress mediates pKr-2-induced neurotoxicity ··· 57 production by microglial NADPH oxidase activation ··· 53
Fig.13. pKr-2induced neuronal cell death in co-culture of cortical microglia and neuron ··· 59
- PART III-
Fig. 14. IL-13/IL-4 induces cell death of microglia activated with pKr-2 ··· 62 Fig. 15. IL-13/IL-4 enhances ROS production via NADPH oxidase in pKr-2
treated microglia ··· 65 Fig. 16. IL-13/IL-4-induced microglia cell death is mediated by ROS (oxidative
stress) through NADPH oxidase ··· 67 Fig. 17. IL-13/IL-4-enhanced COX-2 mediates death of activated microglia by
xiii
ABBREVIATION
6-OHDA, 6-hydroxydopamine
AD, Alzheimer’s disease
ALS,
amyotrophic lateral sclerosisAP, anteroposterior
BV, blood vessel
CAP, capsaicin
CNS, central nervous system
CZP, capsazepine
DA, dopaminergic
CMF-HBSS, Ca2
+-, Mg2
+COX-2, cyclooxygenase-2
-free Hank’s blanced salt solution
CR3, complement receptor type 3
DMSO, dimethyl sulfoxide
DFMO,
α-difluoromethylornithineDTT, dithiothreitol
DV, dorsoventral
DPI, diphenylene iodonium
Eth-1, ethidium homodimer-1
GAPDH, glyceraldehyde-3-phosphate dehydrogenase
GFAP, glial fibrillary acidic protein
xiv
iNOS, inducible nitric oxide synthase
IL-1, interleukin-1
I-RTX, iodo-resiniferatoxin
IL-13, Interleukin-13
IL-4, Interleukin-4
MHC II, major histocompatibility
ML, mediolateral
complex class II
NO, nitric oxide
NeuN, neuron-specific nuclear protein
PCR, polymerase chain reaction
ODC,
Ornithine decarboxylasePD, Parkinson’s disease
PBS, phosphate-buffered saline
PUT, putresine
PMSF, phenylmethylsulfonyl fluoride
ROS, reactive oxygen species
SN, substantia nigra
SD, Sprague-Dawley
SPM, spermine
SPD, spermidine
xv
SNr, Substantia nigra reticulata
TNF-α, tumor necrosis factor
TRPV1,
Transient receptor potential vanilloid subtype 1TH, tyrosine hydroxylase
TUNEL, TdT-mediated dUTP Nick-End Labeling
VTA, ventral tegmental area
1
I. INTRODUCTION
1. Parkinson’s disease (PD)
Parkinson's disease (PD) is the most common chronic progressive neurodegenerative diseases caused by degeneration of neuromelanin-containing dopaminergic neurons in the substantia nigra pars compact (SNpc) and depletion of dopamine in the striatum (STR) (Chana P 2009). In 1817, James Parkinson firstly described the disease in his “An essay on the Shaking Palsy”. Subsequently, the disease was termed as Parkinson’s disease. PD show symptoms such as tremor, or trembling in hands, arms, legs, jaw, and face, rigidity (stiffness in the muscle), bradykinesia (slowness of movement) and impaired balance and coordination (Pahwa et al., 2010; Matthew et al., 2010). Damaged dopaminergic neurons in the SN frequently contain eosinophilic inclusion bodies called Lewy bodies (LBs) (Wakabayashi et al., 2008; Samaranch et al., 2010). Research on the pathogenesis of PD has rapidly advanced due to the development of animal models. Early models were developed by using specific dopaminergic neurotoxins that selectively disrupt or destroy catecholaminergic systems, such as 6-hydroxydopamine (6-OHDA) and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) (Dauer and Przedborski 2003). Recently, it was discovered that agricultural chemicals, such as rotenone, and paraquat, when administered systemically, can also induce specific features of PD (Betarbet et al. 2007). However, the cause of this chronic nigral cell death in PD and its underlying mechanisms remains elusive.
2 2. Cell death of dopaminergic neurons
Oxidative stress is one of the pathogenic factors to be seriously considered as possibility contributing to the loss of dopaminergic neurons in PD (Rajasankar et al., 2009). Elevated levels of reactive oxygen species (ROS) is evident from increased lipid peroxidation and DNA damage in the substantia nigra and increased protein oxidation in the brain of PD (Keeney et al., 2006). The increase in ROS promotes oxidative damage and mitochondria dysfunction lead to neuronal cell death (Abou-Sleiman et al., 2006). Mitochondrial dysfunction in PD has been identified as a systemic deficiency of the electron transport chain Complex I activity. Mitochondrial complex I inhibition has been shown to increase ROS production and subsequently increased oxidative stress. Oxidative stress and mitochondria impairment and this could lead to the accumulation of abnormal protein (Thomas et al., 2007). Intracellular and extracellular aggregates of abnormal protein are a common neuropathological feature. LBs are intracytoplasmic filamentous inclusions that accumulate primarily in subcortical neurons of patients with PD (Wakabayashi et al., 2008; Samaranch et al., 2010). The detection of elevated levels of proinflammatory cytokines in postmortem PD brains has lent strong support to the notion of microglial involvement in the dopaminergic neuron degenerative process (Tansey et al., 2010). Increase of proinflammatory cytokine levels in postmortem PD brains compared to control subjects. Those include pro-inflammatory cytokines (TNF-α, IL-1β, Il-6) (Sugama et al., 2009).
3
The polyamines (putrescine, spermidine and spermine) are aliphatic polycations occurring in all living cells which may affect cell growth and proliferation and in the synthesis of proteins and nucleic acids (Thomas et al., 2001). Polyamines are positively charged and interact electrostatically with polyanionic macromolecules within cells. Spermidine and spermine can bridge the major and minor grooves of DNA, acting as a clamp holding together either two different molecules or two distant parts of the same molecule (Matthews et al., 1993). Polyamines have been implicated in the regulation of several membrane-bound enzymes, including adenylate cyclase, tissue transglutaminase and some ion channels such as N-methyl-D-aspartate (NMDA) (Oliveria et al., 2007), inwardly rectifying K+ (KIR) (Kucheryavykh et al.,2007) and voltage-activated Ca2+ channels (Drouin et al., 1994). Polyamines have also been implicated in the repair of the extracellular matrix, certain signalling processes and cell adhesion. Depletion of polyamines leads to inhibit cell proliferation and migration, or cause defective embryo development, whereas over-accumulation of polyamines induces apoptosis and cellular transformation (Bachrach et al., 2001).
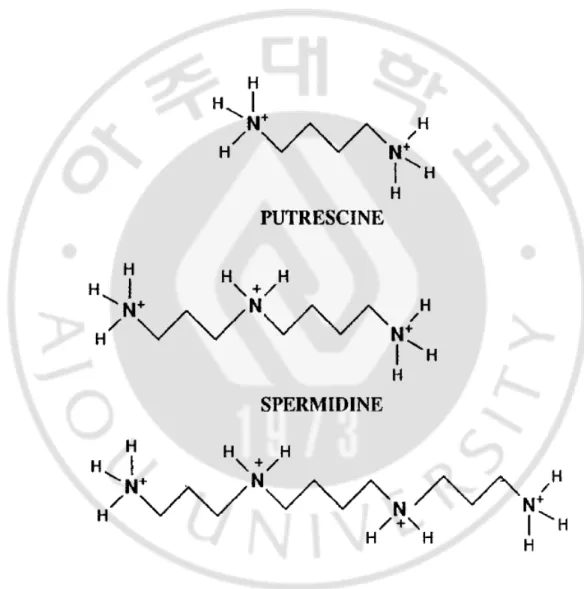
3.1.Structure of polyamines
The polyamine, is an orgnic compound having two or more primary amino group-NH2. Putrescine [1,4-butane diamine or tetramethylenediamine], Spermidine [N-(3-aminopropyl)-1,4-butane diamine or aminopropyl-tetramethylenediamine], Spermine [N,N’-bis(3-aminopropyl)-1,4-butane diamine or diaminopropyl –tetramethyl enediamine] (Fig.1). The functions of the polyamines count on their electrical charg.
4
Polyamines binding energy decreases from spermine to spermidine, and it has been shown, that spermine is the most active and putrescine the least active in the control of various biological process (Yuan et al., 2001).
5 4.Ornithine decarboxylase (ODC)
Ornithine decarboxylase (ODC; ornithine carboxylase, EC 4.1.1.17) is the first enzyme in the synthesis of endogenous polyamines. ODC is in itself an interesting enzyme with several novel regulatory features. ODC activity is highly induced by several CNS injuries (Babu et al., 2003). It has a short half-life (10 min–1 hour) compared with many mammalian enzymes whose half-lives are more often expressed in days (Heby et al., 1985). Increases in ODC activity are one of the early changes observed in cells stimulated to grow and these increases precede changes in DNA synthesis by several hours, making ODC an ‘immediate early’ response gene (Laitinen et al., 1994). ODC is subject to both positive and negative feedback regulation by polyamines, high polyamine concentrations decrease and low polyamine concentrations increase activity. The feedback regulation appears to be a mixture of post-transcriptional regulation and the induction of a unique ODC-specific inhibitor termed ‘antizyme’ (AZ) (Hayashi et al., 1995).
5. Inhibitor of Poyamines metabolism: DFMO
One of the effective inhibitors for polyamine biosynthesis is a specific and active- site inhibitor ODC, α-difluoromethylornithine (DFMO) (Rounbehlerv et al., 2009). DFMO was designed anti-proliferative drug for depleting polyamines from cancer cells. DFMO induced growth arrest and decrease intracellular polyamines in normal and malignant cells (Thomas et al., 2001). Recently, several studies have shown DFMO inhibits apoptosis in ischemia models (Tantini et al., 2006), irradiation-induced models (Deng et
6
al., 2005), TNF-α stimulated chondrocytes (Muscari et al., 2005), and cerebral cortical neuron (Vera et al., 2008).
6. Polyamines and the regulation of cell death
Polyamines are linked in the process of apoptosis (Nitta et al., 2002). The effects of the polyamines are, however, far from simple, with both induction and inhibition of biosynthetic and catabolic enzyme activities being associated with increased and decreased apoptosis (Schipper et al., 2000). In HL-60 cells spermine triggers cytochrome c release from mitochondria, initiates caspase 3 activity and causes cell death through apoptosis (Stefanelli et al., 1999). Apoptosis, induced by TNF-α and cycloheximide, is delayed by the depletion of polyamines in rat IEC and mouse thyroid cells (Claudio et al., 2005). In some cell lines, depletion of polyamines either increases or decreases the sensitivity to apoptosis depending on the nature of apoptotic stimuli. In a cell-free model using a postnuclear extract from U937 cells, the addition of spermine triggers the onset of caspase activity in the presence of ATP and mitochondria or cytochrome c (Stefanelli et al., 2002). The induction of spermidine in L1210 cells overexpressing ODC induces apoptosis (Poulin et al., 1995). The effect of polyamine depletion on apoptosis is different depending on the cell type.
7. Polyamines in PD
Many studies indicate that polyamines spermidine and spermine, and the diamine putrescine may be correlated to cell death (Cetrullo et al., 2010; Hughes et al., 2003).
7
Regarding this, PD and amyotrophic lateral sclerosis (ALS) are associated with an increase in the amount of polyamines in red blood cells (RBC) (Gomes-Trolin et al., 2002). And also, The cellular polyamines putrescine, spermidine, and spermine accelerate the aggregation and fibrillization of α-synuclein, the major protein component of LBs associated with PD (Antony et al., 2003). α -Synuclein is a component of LBs, the morphological hallmark of PD (Takahashi and Wakabayashi, 2001). Traditionally, asynuclein was thought to directly exert damage to DA neurons. Additionally, α-synuclein fails to show dopamine neurotoxicity in microglia-depleted cultures at low concentrations (Zhang et al., 2005), indicating that α-synuclein is also a dual mode toxin. Collectively, these results raise the possibility that under pathological conditions, polyamines may serve as cytotoxic factors in the PD.
8. Charater of TRPV1
Transient receptor potential vanilloid subtype 1 (TRPV1; also known as VR1) belongs to the TRPV subfamily of the large TRP (transient receptor potential) ion channel super family, is a nonselective cation channel that may be activated by the vanilloids such as capsaicin (CAP) and endogenous ligands such as anandamide (AEA) or N arachidonoyl-dopamine (Tominaga et al., 2010). TRPV1 has six transmembrane (TM) domains and a short, pore-forming hydrophobic stretch between the fifth and sixth TM domains (Ferrer-Montiel et al., 2004; Schindl R et al., 2007). TRPV1 are expressed not only in DRG sensory neuron but also many other tissues including the central nervous system (Roberts et al., 2004; Kim et al.,2006; Chen et al., 2009). Also, TRPV1 are
8
produced and expressed in non-neuronal cells, including the squamous cell carcinoma of the human tongue (Marincsák et al., 2009), esophageal epithelial cells (Ma et al., 2010). Activation of TRPV1 induces the accumulation of intracellular Ca2+ and mitochondrial disruption, resulting in cell death (Shin et al., 2003; Sappington et al 2009).
9. Activation of TRPV1 in dopaminergic neuron
Intranigral injection of the TRPV1 agonist CAP into the rat brain, or treatment of rat mesencephalic cultures with CAP, resulted in cell death of DA neurons. This in vivo and in vitro effect was reduced by the TRPV1 antagonist capsazepine (CZP) or iodoresiniferatoxin, suggesting the direct involvement of TRPV1 in dopaminergic neuononal cell death (Kim et al., 2005). Therefore, it is likely that TRPV1 contribute to cell death of dopamine neurons in the substansia nigra in response to the local release of endogenous ligands such as polyamines.
10. Polyamines are endogenous ligand for TRPV1
Polyamines are positively charged and thus can interact electrostatically with negatively charged nucleic acids and proteins, including ion channels. TRPV1 is regulated by protons (Tominaga et al., 2010) and by extracellular cations including Na+, Mg2+, and Ca2+ (Ahern et al., 2005). Extracellular spermine, spermidine, and putrescine sensitize TRPV1 receptor in sensory neurons (Fig. 2). Spermine activates TRPV1 and increases capsaicin-evoked currents (Ahern et al., 2006). Polyamines
9
regulates TRPV1 in a charge-dependent manner (spermine, 4+ > spermidine, 3+>> putrescine, 2+) (Ahern et al., 2006). Polyamines are important endogenous ligand of TRPV1. Therefore, it is likely that polyamines as endogenous ligand activate TRPV1 receptor in dopaminegic neurons.
11. Prothrombin Kringle-2 (pKr-2)
Kringle, a structure with three characteristic intradisulfide bonds, is found in a number of proteins, including prothrombin, plasminogen, hepatocyte growth factor (HGF), macrophage-stimulating protein (MSP) and apolipoprotein. Kringles of individual proteins have distinct functions in many cell types. Prothrombin, a precursor of thrombin, is converted to an amino terminal fragment, designated kringle-1-2, and active thrombin by the prothrombinase complex (Factor Xa) (Shikamoto and Morita, 1999), in turn, inducing blood coagulation (Davie et al., 1991). Thrombin formed during activation further cleaves kringle-1-2 into kringle-1, including the Gla domain, a kringle-1 domain and kringle-2 (Mann, 1976; Taneda et al., 1994). Prothrombin kringle-2 (pKr-2) inhibits basic fibroblast growth factor (bFGF)-stimulated bovine capillary endothelial cell growth (Kim et al., 2005), angiogenesis in the chorioallantoic membrane of chick embryos (Lee et al., 1998; Rhim et al., 1998), procoagulant activity of newly formed thrombin (Dasgupta and Thiagarajan, 2007), and Lewis lung carcinoma (LLC) tumor growth and metastases in C57BL6/J mice lungs through anti-angiogenic effects (Kim et al., 2006; Kim et al., 2005). Prothrombin kringle-2 (pKr-2) is generated from the precursor prothrombin endogenously expressed in human, mouse,
10
and rat brain, including the cortex (Dihanich et al., 1991; Soifer et al., 1994; Weinstein et al., 1995).
Fig.2. Relative activation and permeation of TRPV1 by polyamines. A, structures of the, putrescine, spermidine, and, spermine. B, summary of currents produced by 5-µM concentrations of spermine, spermidine, and putrescine in HEK293 cells. C, current-voltage relationship for responses produced by 30-µM concentrations of either spermine or spermidine with the pipette solution containing 150 µM NaCl. D, current-voltage relationship for responses evoked by 30 nM capsaicin (in Na+ medium) and 30 mM putrescine_capsaicin (bold line) (Ahern et al., 2006).
11 12. Microglia
12.1. Features of microglia
Ten percent of glial cells are microglia in the mature brain. Microglial cells differentiate in the bone marrow from hematopoietic stem cells, the progenitors of all blood cells. Microglia is the resident macrophages of the brain and spinal cord, and thus acts as the first and main form of immune response and maintaing homeostasis in the central nervous system (CNS). While microglia are constantly moving through the CNS, if the microglia cell find any infectious agents, damaged neurons, neural tangle microglia will activae (Mandrekar-Colucci et a., 2010). Microglia must react quickly to destroy the infectious agents before they damage the sensitive neural tissue. Microglia changes their morphology in response to brain injury (Perry et al., 2010). There are marked variation in the expression of membranous markers and in the biochemical activities between the inactivated microglia (ramified) and the activated microglia (Ameboid and Round) (Perry et al., 2010).
12.2. Involvement of microglial activation in neurodegerative diseases
Evidence from postmortem brain analysis implicates the activated microglia in progressive neurodegenerative disorders (Luo et al., 2010). Activated microglia releases numerous of proinflammatory cytokines and potentially neurotoxic mediators such as TNF-α, PGE2 (Gong et al., 2010) and IFN-r and oxidative stress (Block et al., 2006) that may contributes to the neuronal damage. Reduction of these cytokines shows that significant reduction cell death of hippocampal neuron after ischemia (Xiong M et al., 2009). Some cytokines such as IL-1 and TNF-α induce microglia to produce nitric
12
oxide (NO) and further damage neurons through lipid hyper-peroxidation and DNA injury. This demonstrates that cytokines are directly involved in neuronal death. The chronic inflammation associated with neurodegenerative disease is to be found in studies on AD and PD (Tansey et al., 2010; Mandrekar-Colucci et al., 2010). In pathologically vulnerable regions of the AD brain reveals that large large numbers of activated microglia associated with the neuronal death. Activated microglia reportedly play a major role in the development of gliosis in postmortem PD brains (Lee et al., 2010), it is believed that proinflammatory cytokines are directly involved in gliosis formation. In animal models of nigrostriatal degeneration using 6-OHDA and MPTP studies have shown that proinflammtory cytokines secreted in activated microglia contribute to neuronal degeneration. These several studies show possible involvement of activated microglia in the initiation and/or progression of neuropathological processes of several neurodegenerative diseases such as AD and PD.
13. Apoptosis of activated microglia
Microglial cells play an important role in brain inflammatory responses. Microglia responses normally to neuronal damage and remove the damaged cells and infectious agents by phagocytosis before they damage the sensitive neural tissue (Colton et al., 2010). Persistent activation of microglia in the brain contributes to
irreversible tissue damage associated with many age-related chronic diseases through
the release of potentially cytotoxic molecules such as proinfalmmatory cytokines, reactive oxygen species, proteinases and complement proteins in chronic
13
neurodegenerative diseases (Luo et al., 2010; Brown et al., 2010). The control mechanism to suppress inflammatory responses is importance to suspension of chronic microglia activation in various neurodegenerative diseases. Previous studies have shown that the death of activated microglia lead to the termination of the process of brain inflammation (White et al., 1998; Yang et al., 2006), which induce the neuronal degeneration. Microglia cell death could be induced by injection of toxins such as a mitochondrial toxin 3-nitropropionic acid (3-NP) (Ryu et al., 2003), 6-OHDA (Takai et al., 1998), or LPS (Shin et al ., 2004). Also, oxidative stress from activated microglia induced by stimulation agents such as menadione (Hollensworth et al., 2000), chromogranin A (Kingham et al., 2000), or LPS (Lee et al., 2001; Liu et al., 2001) is contributes to death of microglia. Augmenting the death of activated microglia can be included in the strategy for restoration of balanced immune responses during aging and neurodegenerative diseases.
14. Interleukin-13 (IL-13), Interleukin-4 (IL-4)
The cytokines IL-13 and IL-4 are produced by Th2 cells (Wynn, 2003; Nelms et al., 1999), and recruit and activate IgE-producing B cells, and enhance IgE mediated responses (Foster et al., 2002; Mueller et al., 2002). IL-13 and IL-4 are cytokines critical to the development of T cell-mediated humoral immune responses, which are associated with allergy and asthma, and exert their actions through three different combinations of shared receptors. For signal transduction, both IL-4 and IL-13 require the same receptor subunit, IL-4Ra (Wynn, 2003). Receptor sharing by these two
14
cytokines is the molecular basis for their overlapping biological functions (LaPorte et al., 2008). The biological effects of IL-13/IL-4 are related to a single transcription factor, signal transducer and activator of transcription 6 (STAT6) (LaPorte et al., 2008). Interleukin-13 (IL-13), Interleukin-4 (IL-4) can down-regulate the production of inflammatory mediators from activated microglia and macrophages thus providing negative feedback (Ledeboer et al., 2000; Burmeister et al., 2009; Fukuyama et al., 2009). In the CNS, IL-13/IL-4 decrease the secretion of proinflammatory cytokines such as TNF-α, iNOS, cyclooxygenase (COX)-2 from activated microglia in vivo and in vitro (Ledeboer et al 2000; Furlan et al., 2000; Liu et al., 2010). IL-13 contributed to survival of mice via significantly reduction of the secretion of cytotoxic molecules (Matsukawa et al., 2000). Also, IL-4 was shown to attenuate cell death in an animal model of multiple sclerosis (MS) through the downregulation of inflammatory mediators (Furlan et al., 2000). Furthermore, previous studies reported that IL-13/IL-4 induces the death of activated rat microglia via downregulation of inflammatory mediators in vitro (Yang et al., 2002; Yang et al., 2006). IL-13/IL-4 contributed to that is responsible for control of brain inflammation. Activated micoglia cell death is important for neuronal defense systems, because they are part of the integral inflammatory and anti-inflammatory systems.
15 15. Aims of study
PART I.
(1) Specific aims of this study was to investigate whether DFMO altered 6-OHDA induced neurotoxicity of dopaminergic neurons in vivo and invitro.
(2) To investigated whether 6-OHDA/polyamine-induced cell death of dopaminergic neuron was mediated by activation of TRPV1 in dopaminergic neurons
(3) To investigated whether 6-OHDA-induced activation of AKT, caspase-3 in mesencephalic cultures could be affected by an irreversible and specific inhibitor of ODC, DFMO.
PART II.
(1) To investigate whether the effects of prothrombin kringle-2 on microglia-mediated immune/inflammatory process participate in the neurodegeneraion in cortex
PART III.
(1) To investigate whether the IL-13/IL-4 enhnces ROS via activation of NADPH oxidase in Prothrombin kringle-2-treated microglia culture.
(2) To investigate whether the IL-13/IL-4 induces cell death of microglia in Prothrombin kringle-2-treated microglia through NADPH oxidase derived ROS and COX-2.
16
II. MATERIALS AND METHODS
A. METHODS
1. Sterotaxic surgery and drug injection
All experiments were carried out in accordance with approved animal protocols and guidelines established by Ajou University. Female Sprague-Dawley (SD) rats (260-280g) were anesthetized by injection of chloral hydrate (360 mg/kg, i.p.) and positioned in a stereotaxic apparatus (David Kopf Instrument, Tujunga, CA, USA). A midline sagittal incision was made in the scalp, and holes were drilled in the skull over the lateral ventricles and dorsal cortex using the following coordinates: 0.8 mm posterior to bregma and 1.5 mm lateral to the midline for intracerebroventricular (i.c.v) injections; and 1.4 mm posterior to bregma and 2.0 mm lateral to the midline for intracortical injections according to Paxinos and Watson (1998). The hole of the tip was directed vertically down to 3.6 mm beneath the surface of the brain for the ventricles and to 2.0 mm for cortex. All injections were made using a Hamilton syringe equipped with a 30S-gauge beveled needle and attached to a syringe pump (KD Scientific, New Hope, PA, USA). Infusions were made at a rate of 0.2 µl/min for pKr-2 (48 µg/4µl in sterile phosphate-buffered saline [PBS]; Haematologic Technologies Inc), and 0.5µl/min for diphenylene iodonium (DPI; 100 μM in 20 µl sterile saline; Calbiochem), and for vehicle (PBS) as controls.
17
Animals were anesthetized with chloral hydrate (360 mg/kg, i.p.) at the indicated time points after injection and transcardially perfused with saline solution containing 0.5% sodium nitrate and heparin (10 U/ml), followed by fixation with 4% paraformaldehyde dissolved in 0.1 M phosphate buffer (PB). Brains were removed from the skull, postfixed overnight at 4°C in buffered 4% paraformaldehyde, and stored at 4°C in 30% sucrose solution until they sank. Brains were frozen sectioned using a sliding microtome into 40 μm coronal sections and collected in six separate series. Immunohistochemistry was performed using the avidin– biotin staining technique as described previously (Choi et al., 2003a; Choi et al., 2003b). Briefly, free-floating serial sections were rinsed three times for 10 min in PBS and then pretreated for 5 min at room temperature in PBS containing 1%H2O2. Sections were then rinsed in PBS containing 0.3% Triton X-100 and 0.5% BSA and then preincubated for 1 hour at room temperature in PBS containing 0.5% BSA. Next, the sections were incubated overnight with gentle shaking at room temperature with PBS containing 0.5% BSA and the following monoclonal primary antibodies: anti-OX-42 (specific for complement receptor type 3 (CR3); Serotec), anti-OX-6 (specific for major histocompatibility complex class II antigens; Pharmingen), anti-ED1 (specific for glycosylated lysosomal antigen; Serotec) for microglia, anti-neuron-specific nuclearprotein (NeuN; Chemicon) for neurons. Sections were then rinsed in PBS and incubated for 1 hour at room temperature in 1:200 biotin-conjugated anti-mouse antibody in PBS containing 0.5% BSA. Sections were rinsed again and incubated for 1
18
hour at room temperature in avidin– biotin complex solution (Vector Laboratories, Burlingame, CA). After rinsing three times in PBS, the signal was detected by incubating sections in 0.5 mg/ml 3,3 diaminobenzidine in PB containing 0.003%H2O2. Sections were then rinsed in PBS, mounted on gelatin-coated slides, and viewed under a bright-field microscope (Olympus Optical, Tokyo, Japan). For Nissl staining, some of the cortex tissue was mounted on gelatin-coated slides, dried for 1 hour at room temperature, stained with 0.5% cresyl violet (Sigma), dehydrated, coverslipped, and then analyzed under a bright-field microscope (Olympus Optical).
3. Double-immunofluorescence staining
For double-immunofluorescence staining, sections were processed as described previously (Choi et al., 2005b). Briefly, sections were mounted on gelatin-coated slides,dried for 6 hours at room temperature, and washed twice in PBS. Slides were incubated overnight at 4°C in a combination of a goat anti-TNF-α (1:100; R&D systems), and goat anti-IL-1β (1:200; R&D system), a rabbit anti-iNOS (1:200; Upstate biotechnology, Lake Placid, NY), and then exposed for 1 hour at room temperature to fluorescein-conjugated lycopersicon esculentum (tomato lectin) (1:200; Vector Laboratories) and Texas-Red-conjugated anti-rabbit IgG (Vector Laboratories) or Texas-Red-conjugated anti-goat IgG (Vector Laboratories). Slides were also incubated overnight at 4°C with anti-p67phox antibody (1:200; BD Biosciences) and OX-42 (1:100; Serotec) or GFAP (1:100; sigma). After washing in PBS, the sections were incubated
19
simultaneously with a mixture of FITC-conjugated goat anti-mouse IgG (1:100; Vector Laboratories) and Texas Red-conjugated goat anti-rabbit IgG (1:100; Vector Laboratories) for 1 hour at room temperature. Slides were coverslipped with Vectashield medium, and viewed using an IX71 confocal laser scanning microscope (Olympus). To analyze the localization of different antigens in double-stained samples, images were obtained from the same area and merged using interactive software.
4. Live and dead assay
Viability of microglia was assessed by double labeling of cells with 2 μM calcein-acetoxymethyl ester (calcein-AM) and 4 μM ethidium homodimer-1 (Etd-1). The calcein–positive live cells (green) and ethidium positive dead cells (red) were counted in eight fields from two different wells for each group using a confocal laser-scanning microscope (Kim et al., 2006).
5. MTT Reduction Assay
As previously described (Park et al., 2008), MTT stock solution was added to each culture after pKr-2 treatment so that the final concentration of MTT in the medium was 0.25 mg/ml (600 μM), and the cells were incubated for 3 hours at 37°
C After incubation, medium was removed, and DMSO was added to each well. The absorbance was measured with a microplate reader at a test wavelength of 570 nm and a reference wavelength of 655 nm.
20
6. TdT-mediated dUTP Nick-EnD Labeling (TUNEL) assay
Cell death effect by a combination pKr-2 and IL-13 was determined using the Apoptag Fluorescein Direct in situ Apoptosis Detection kit (Intergen) that detects the 3′-OH region of cleaved DNA. TUNEL staining was followed by kit protocol. Microglia cultures were treated with pKr-2 (50 μg/ ml) in the absence or presence of IL-13 (20 ng/ ml) for 6 days. And then cultures were fixed in 1% paraformaldehyde in PBS, incubated with a mixture of TdT and reaction buffer containing FITC fluorescein-conjugated- digoxigenin-dUTP (Intergen) in a humidified chamber for 1 h at 37°C, and washed in washing buffer (Intergen). For double staining of TUNEL with anti-OX-42, cultures were incubated with anti-OX-42 Abs for overnight at room temperature, and then applied with Texas Red-conjugated anti-mouse IgG (Vector Laboratories) for 30 min, and viewed using a confocal laser-scanning microscope.
7. Immunocytochemistry
As previously described (Lee et al., 2006), cultured cells were paraformaldehyde (4%) fixed, permeabilized with 0.2% Triton X-100 for 5 min, and then incubated overnight with the anti-mouse OX-42 (specific for complement receptor type 3; Serotec) for microglia. On the following day, cultured cells were rinsed with PBS 1 0.5% BSA and incubated with appropriate biotinylated secondary antibody, followed by avidin-biotin complex (Elite Kit purchased from Vector, Burlingame, CA). The bound antiserum was visualized by incubating with 0.05% diaminobenzidine- HCl (DAB) and 0.003% hydrogen peroxide in 0.1 M PB. The DAB reaction was stopped by rinsing tissues in
21
0.1 M PB. Immunostained cells were analyzed under brightfield microscope.
8. Reverse transcription-polymerase chain reaction (RT-PCR)
Brain tissues from the ipsilateralcortex were dissected at the indicated time points afterinjection, and total RNA was extracted in asingle step using RNAzol B (Tel-Test, Friendswood, TX) following the instructions of the manufacturer. Total RNA was reversetranscribed into cDNA using Superscript II reverse transcriptase(Invitrogen, Rockville, MD) and random primers (Promega, Madison,WI). The primer sequences used in this study were as follows:5'-TGA TGT TCC CAT TAG ACA GC-3' (forward) and 5'-GAG GTG CTGATG TAC CAG TT-3' (reverse) for interleukin-1β (IL-1β); 5'-GCA GAATGT GAC CAT CAT GG-3' (forward) and 5'-ACA ACC TTG GTG TTG AAGGC-3' (reverse) for iNOS; and 5'-TCCCTC AAG ATT GTC AGC AA-3'(forward) and 5'-AGA TCC ACA ACG GAT ACA TT-3' (reverse) forglyceraldehyde phosphate dehydrogenase. The PCR amplificationconsisted of 30 cycles of denaturation at 94°C for 30 s,annealing at 56°C for 30 s (for IL-1β and iNOS), and extension at 72°C for90 s. PCR products were separated by electrophoresis on 1.5% agarose gels, after which the gels were stained with ethidiumbromide and photographed. For semiquantitative analyses, the photographs were scanned using the Computer Imaging Device andaccompanying software (Fujifilm, Tokyo, Japan).
9. Western blot analysis
22
lysis buffer containing the following (in mM): 20 Tris-HCl, pH 7.5, 1 EDTA, 5 MgCl2, 1 dithiothreitol, 0.1 phenylmethylsulfonyl fluoride, and protease inhibitor mixture (Sigma). The tissue homogenates was centrifugedat 4°C for 20 min at 14,000 x g, and the supernatant wastransferred to a fresh tube. The extracts were frozen and keptat -80°C. For subcellular fractionation, protein extractsof both the cytosolic and plasma membrane factions were prepared fromthe ipsilateral cortex at the indicated time points after pKr-2 injection. Tissues were gently homogenized using aglass homogenizer in ice-cold buffer consisting of the following(in mM): 20 HEPES, 250 sucrose, 10 KCl, 1.5 MgCl2, 2 EDTA, and protease inhibitor mixture (Sigma). Homogenates were centrifugedfor 5 min at 500 x g at 4°C, and supernatants were collectedand centrifuged for 20 min at 13,000 x g at 4°C. The pelletswere further centrifuged for 1 hour at 100,000 x g at 4°C,and the resulting supernatants and pellets were designated asthe cytosolic and plasma membrane fractions, respectively. Equal amountsof protein (50 µg) were mixed with loading buffer (0.125M Tris-HCl, pH 6.8, 20% glycerol, 4% SDS, 10% mercaptoethanol, and 0.002% bromophenol blue), boiled for 5 min, and separated by SDS-PAGE. After electrophoresis, proteins were transferred to polyvinylidene difluoride membranes (Millipore, Bedford, MA) using an electrophoretic transfer system (Bio-Rad, Hercules,CA). The membranes were washed with Tris-buffered saline solution(TBS) and then blocked for 1 hour in TBScontaining 5% skim milk. The membranes were then incubated overnightat 4°C with mouse anti-p67phox (1:500;BD Biosciences).After washing, the membranes were incubated for 1
23
hour at room temperature withsecondary antibodies (1:2000; Amersham Biosciences, Arlington Heights, IL) and washed again. Finally, the blots were developed with enhanced chemiluminescence detection reagents (Amersham Biosciences). To determine the relative degree of membrane purification, the membrane and cytosolic fraction was subjected to immunoblotting for calnexin, a membrane marker, using a rabbit polyclonal antibody against calnexin (1:4,000; Santa Cruz Biotechnology). Activation of AKT and caspase-3 was examined by immunoblot analysis with antibodies specific for the phosphorylated forms of AKT (1:1,000) and cleaved caspase-3 (1:1000), respectively. For semiquantitative analyses, the densities of bands on immunoblots were measured with the computer imaging device and accompanying software (Fujifilm). 10. In situ detection of O2 - and O2 - Hydroethidine -derived oxidants
histochemistry was performed for in situ visualization of O2- and O2 -derived oxidants (Choi et al., 2005b; Wu et al., 2003). Forty-eight hoursafter pKr-2 injection, hydroethidine (1 mg/ml in PBS containing1% dimethylsulfoxide; Molecular Probes, Eugene, OR) was administeredintraperitoneally. After 15 min, the animals were transcardially perfused with a saline solution containing 0.5% sodium nitrate and heparin (10 U/ml) and then fixed with 4% paraformaldehydein 0.1M PB. After fixation, the brains were cut into 40 µmslices using a sliding microtome. Sections were mounted on gelatin-coated slides, and the oxidized hydroethidine product, ethidium, wasexamined by IX71 confocal microscopy (Olympus Optical).
24 11. Detection of protein oxidation
The extent of protein oxidation was assessed by measuring protein carbonyl levels with an OxyBlot protein oxidation detection kit (Chemicon, Temecula, CA) accordingto the protocol of the manufacturer as described previously (Choi et al., 2005b). Protein samples were prepared from rat brains harvested 48 hours after injection with pKr-2 in the absence or presence of DPI (100 μM, i.c.v) or trolox (50 mg/kg, i.p.). Subsequently, protein samples (15µg) were mixedin a microcentrifuge tube with 5 µl of 12% SDS and 10 µl of 1x 2,4-dinitrophenylhydrazine (DNPH) solution. Ten microliters of 1x neutralization solution (a kit component)was added instead of the DNPH solution as the negative control.Tubes were incubated at room temperature for 15 min and then mixed with 7.5µl of neutralization solution. Next, the samples weremixed in equal volumes of SDS sample buffer and separated by SDS-PAGE. After electrophoresis, proteins were transferred topolyvinylidene difluoride membranes (Millipore). The membranes were then blocked for 1 hour at room temperature in TBS containing 0.1% Tween 20 and 1% BSA. Membranes were incubated overnightat room temperature with the anti-DNPH antibody (1:150) and then incubatedat room temperature for 1 hour with secondary antibodies (1:300). Blots weredeveloped using enhanced chemiluminescence reagents (AmershamBiosciences). Proteins that underwent oxidative modification(i.e., carbonyl group formation) were identified as a band inthe samples derivatized with DNPH. The optical density of
25
the bands was measured using the Computer Imaging Device and accompanying software (Fujifilm). Levels of protein carbonyls were quantifiedand expressed as the fold increase versus untreated controls.
12. Measurement of ROS Generation
For measurement of intracellular ROS levels, cells were incubated with 10 μM 5- (and 6-) chloromethyl-20,70-dichlorodihydro-fluorescein diacetate [CM-H2DCF-DA (here referred to as DCF)] (Invitrogen, San Diego, CA) for 10 min. The cells were then washed with D-PBS (in mM: 2.68 KCl, 1.47 KH2PO4, 136.89 NaCl, and 8.1 Na2HPO4), and the fluorescence images were taken with a IX71 confocal laser microscope (Olympus). Extracellular superoxide (O2•_) production from microglia was determined as reported previously by measuring the superoxide dismutase (SOD)-inhibitable reduction of tetrazolium salt, WST-1. In brief, immediately before treatment, enriched microglia were washed twice with Hanks’balanced salt solution (HBSS). To each well, 100 μl of HBSS with or without SOD (600 U/ml), 50 μl of vehicle, or, and 50 μl of WST-1 (1 mM) in HBSS were added. The cultures were incubated for 30 min at 37°C, and the absorbance at 450 nm was read with a SpectraMax Plus microplate spectrophotometer (Molecular Devices, Sunnyvale, CA, U.S.A.). The amount of SOD-inhibitable (O2•_) was calculated and expressed as percentage of vehicle-treated control cultures. Cell-free experiments determined that PQ treatment alone did not affect absorbance.
26
13. Primary culture of cortical neuron, drug treatement, and assessment neuronal cell death
Rat cortical neuronal cultures were prepared from the 17-day-old fetal brain, and the neocortices were mechanically triturated as described previously (Lee da et al., 2006). Dissociated cells were plated on 12 mm round aclar plastic coverslips in 24-well plates and Ara C (2µM) was added at 2–3 days in vitro (DIV 2–3). At DIV 6, cells were treated with pKr-2 or PBS in MEM containing 2% B-27 supplement, 21mM glucose, 26.5mM sodium bicarbonate, 2mM L-glutamine, and penicillin–streptomycine deprived of serum for 24 hours.
14. Primary culture of dopaminergic neuron, drug treatement, and assessment neuronal cell death
Neuron-enriched mesencephalic cultures were prepared as previously described. In brief, cells from the ventral mesencephalons of embryonic day 14 Sprague–Dawley rats were seeded on 12 mm round aclar plastic coverslips or culture plates pre-coated with poly-D-lysine (Sigma, St. Louis, MO) and laminin (Upsatate Biotech, NY) at a density of 1.25–2.0 · 105 cells/cm. The cultures were incubated in a humidified incubator at 37°C, 5% CO2 for 24 h. To suppress the proliferation of glial cells, on the
second day in vitro (DIV2), media were replaced with a chemically defined serum-free media (DM) composed of Ham’s nutrient mixture F12/DMEM and supplemented with 1% ITS (insulin, transferrin, and sodium selenite), glucose, L-glutamine, and P/S. On
27
DIV4, cultures were treated with various stimulus (Matreya, Pleasant Gap, PA) in DM without ITS. At the time cultures were immunostained with cell-type specific antibodies (see below: Immunocytochemistry), the cell composition included 5% astrocytes and less than 1% microglia. The remaining cells were presumed to be neurons, 4.5–6% and 9.5–11% of which were tyrosine hyroxylase-immunopositive (TH-ip) and gamma amino butyric acid-immunopositive (GABA-ip) neurons, respectively
15. Cortical Microglia Cultures
Microglia were cultured from the cerebral cortices as described previously (Lee da et al., 2006). Briefly, cerebral cortices were isolated from postnatal 1 day SD rats, and meninges and blood vessels were removed. Tissues were triturated and plated in 75-cm2 T-flasks. Cells were grown for 2 weeks in 75-cm2 T-flasks (0.5 hemisphere/flask). The microglia were then detached from the flasks by mild shaking and filtered through a nylon mesh to remove astrocytes and clumped cells. Cells were plated into 24-well plates (1 × 105 cells/well) or 35-mm culture dishes (5 × 105 cells/well). After 1 hour, the culture medium was changed to MEM containing 5% FBS.
16. Co-cultures of microglia and neurons
At DIV 5, neuron-enriched cortical cultures plated on 12-mm round aclar plastic Cover slips (1 × 105
28 × 105
cortical microglia/well. After 1 hour, the culture medium was replaced with neuronal culture medium (MEM medium containing 5% FBS); Twenty-four hours later, the co-cultures were treated with pKr-2 (50 µg/ml) for 24 hours and were processed for immunocytochemistry.
17. ODC enzyme activity assay
Neuronal culture plates were placed on ice and washed with PBS and scraped in a buffer consisting of 0.1 mM EDTA, 0.02 mM pyridoxal phosphate, 2.5 mM dithiothreitol in 10 mM sodium phosphate buffer, pH 7.2. The homogenate/lysate was centrifuged at 11,000 rpm for 15 min. ODC activity in the supernatant was measured by determining the amount of 14C–CO2 from L-1-14C-ornithine at 37°C during a 60 min incubation. One unit of enzyme activity corresponds to 1 nmol of decarboxylated substrate/h of incubation.
18. Dopamine (DA) uptake assay
The measurement of dopamine uptake was performed as described previously (Lee da et a., 2006). Briefly, cultures were washed twice with the incubation solution (HBSS containing 10 mM Hepes, 0.6% glucose, 0.2 mM pargyline, and 0.01% ascorbic acid, pH 7.4), and then incubated at 37°C for 20 min in incubation solution with a final concentration of 83.3 nM tritiated dopamine ([3H]DA, 444 GBq/mmol; Amersham, Oakville, ON). Blank values were obtained by incubating cultures at 0°C. The reaction was terminated by removal of the solution followed by three rapid washes with ice-cold
29
incubation solution. The cultures were then lysed with lysis buffer (0.2M NaOH containing 0.2% Triton X-100) and transferred to scintillation vials for counting.
19. Measurement of the densities of the immunoblot bands
For semiquantitative analysis of immunoblot bands, the density of each band was measured with a computer imaging device and accompanying software (Bio-Rad).The background value was subtracted from all other readings
20. Determination of NO
As previously described (Lee et al., 2005; Yang et al., 2002), the amount of nitrite formed from nitric oxide was measured by mixing the culture medium (50 µl) with an equal volume of Griess reagent (0.1% naphthylethylene diamine, 1% sulfanylamide and 2.5% H3PO4). The optical density was measured at 450 nm.
21. Quantification of TNF-α Release
The release of TNF-α into the culture supernatant was determined by enzyme-linked immunosorbent assay (ELISA) (Lee et al., 2005). Supernatants (50 µl) were placed on ELISA kit strips (Biosource). Sandwich ELISA was then performed according to the manufacturer's instructions.
22. Statistical analysis
All values are expressed as mean ±SEM. Statistical significance (p < 0.05 for all analyses)was assessed by ANOVA using Instat 3.05 (GraphPad Software,San Diego,
30 CA), followed by Student-Newman-Keuls analyses.
31
III.RESULTS
Part I. DFMO, Ornithine Decarboxylase Inhibitor, Prevents 6 hydroxydopamine-Induced Neurotoxicity in Rat Mesencephalic Dopaminergic Neuron
1.
Polyamine depletion protects on 6-OHDA-induced neurotoxicity of dopaminergic neurons in neuron-enriched mesencephalic cultureMesencephalic cultures were treated with 6-hydroxydopamine (6-OHDA) (30 µM) (Fig. 3B) for 24 hours and processed for immunostaining with anti-TH to detect dopaminergic neurons. TH immunocytochemical staining demonstrated that the dopaminergic neurons in control cultures had large, healthy soma with long, branched neuritic processes (Fig. 3A). In contrast, treatment with 6-OHDA (30 µM) (Fig. 3B) produced a profound loss of dopaminergic neurons. Moreover, the remaining TH-ip neurons had an injured morphology, with rounded and shrunken cell bodies and short processes. To investigate whether DFMO altered 6-OHDA induced neurotoxicity of dopaminergic neurons in neuron-enriched mesencephalic culture, DFMO was pretreated for 30min, before 6-OHDA treatement. The results of TH immunohistochemistry showed that DFMO treatment significantly inhibited 6-OHDA-induced neuronal death in neuron-enriched mesencephalic cultureand relatively healthy dopaminergic neurons with branched processes (Fig. 3C). Consistent with these findings, counting of TH-ip neurons and the levels of [3H] dopamine uptake were measured in cultures treated with 6-OHDA (30 µM) or 6-OHDA in the presence of
32
DFMO for 24 hours. When quantified and expressed as a percentage of control values, treatment with 6-OHDA (30 µM) attenuated the number of TH-ip neurons and the level of [3H] dopamine uptake by 31–47% and 27–56%, respectively, compared with untreated control cultures (Fig. 3D). however, DFMO treatment inhibites 6-OHDA-induced neuronal death and increased the level of [3H] dopamine uptake in neuron-enriched mesencephalic culture (Fig.3 D).
34
Fig. 3. Effects of DFMO, ornithine decarboxylase on 6-hyroxydopmaine (6-OHDA)-induced neurotoxicity of dopaminergic neurons in neuron-enriched mesencephalic culture. At 4 days in vitro (DIV 4), neuron-enriched mesencephalic culture were untreated (A) or treated with 6-OHDA (30 μM) in the absence (B) or presence of DFMO (500 μM) (C) for 24 hours and immunostainined with TH-antibody. (C), DFMO blocks 6-OHDA-induced cell death of dopaminergic neurons in mesencephalic cultures. (D), The number of TH-ip neurons was counted or the levels of [3H] dopamine uptake were measured in cultures treated with 6-OHDA (30 μM) or 6-OHDA in the presence of DFMO for 24 hours. (E) Effects of various doses of DFMO protects TH-ip neurons from 6-OHDA toxicity in mesencephalic cultures. Graph represents the mean ± SEM of triplicate cultures for five separate platings expressed as a percentage of the control. *P < 0.001 compared with vehicle, #P < 0.001 compared with 6-OHDA alone treated cultures (ANOVA and Student-Newman-Keuls analyses).
35
2. ODC activity in neuron-enriched mesencephalic culture exposed to 6-hydroxydopamine (6-OHDA)
I examined effect of ODC ativity in mesencephalic dopaminerguc neuron culture treated with 6-OHDA in the absence or presence of DFMO (500 μM) (Fig. 4). I found that in mesencephalic dopaminergic neuron cultures increased after 90 min of treated 6-OHDA (30 μM). Following this early induction, ODC activity returned to control levels within 24 hours. The induction of ODC activity was completely prevented when the cells were pretreated with DFMO (500 μM), an irreversible and specific inhibitor of ODC.
36
Fig. 4. ODC activity in dopaminergic neurons exposed to 6-hyroxydopmaine (6-OHDA). Ddopaminergic neurons were grown for 24 hours after plating in the presence or absence of 500 μM DFMO, afterward the cells were exposed to 6-OHDA (30 μM). The cells were collected at the indicated time and analyzed for ODC activity. Graph represents the mean ± SEM of triplicate cultures for five separate platings expressed as a percentage of the control. *P < 0.001 compared with control cultures (ANOVA and Student-Newman-Keuls analyses).
37
3. Spermidine and Spermine induces neurotoxicity of dopaminergic neurons in neuron-enriched mesencephalic culture
I examined the cytotoxic effect of Putresine (PUT; 100 µM), Spermidine (SPD;100 µM), and Spermine (SPM;100 µM) on DA neurons by determining the number of TH-positive neurons and the rate of [3H]dopamine uptake. PUT (100 µM), SPD (100 µM), and SPM (100 µM) were treated for 24 hours in mesencephalic cultures. Compared with non-treated control, SPD (100 µM) and SPM (100 µM) exhibit neurotoxicity in mesencephalic cultures. PUT (100 µM) failed to exhibit neurotoxicity against TH-ip neurons in mesencephalic cultures (Fig. 5A). Spermidine and Spermine-induced neurotoxicity occurred in a dosedependent manner, as determined by the number of TH-positive neurons (Fig. 5B,C).
38
Fig. 5. Spermidine (SPD) and Spermine (SPM) induces cell death of dopaminergic neurons in mesencephalic cultures. Dose-dependent neurotoxicity of SPD (B) and SPM (C). Cell viability was determined by counting the number of TH-ip neurons. Graph represents the mean ± SEM of triplicate cultures for five separate platings expressed as a percentage of the control. *P < 0.05, **P < 0.001 compared with vehicle (ANOVA and Student-Newman-Keuls analyses).
39
4.Polyamines and 6-OHDA induces loss of dopaminergic neurons through TRPV1 in neuron-enriched mesencephalic culture
Recently, I reported that activation of TRPV1 induces cell death of dopaminergic neuron in vivo and in vitro (Kim et al., 2005). In addition, other groups reported that endogenous polyamines are Potent Ligand for Capsacin Receptor TRPV1 (Ahern et al., 2006). I therefore investigated whether 6-OHDA-induced cell death of dopaminergic neuron was mediated by activation of TRPV1 in neuron-enriched mesencephalic cultures in vitro. Pretreatment of mesencephalic cultures with CZP (10 µM) or CZP (30 µM) attenuated the effects of 6-OHDA (30 µM), indicating TRPV1-mediated neurotoxicity. I also tested sperimidine or spermine-induced neurotoxicity was mediated by activation of TRPV1 in neuron-enriched mesencephalic cultures in vitro. Pretreatment of mesencephalic cultures with CZP partially attenuated the effects of spermidine (50 µM), Spermine (50 µM) indicating TRPV1-mediated neurotoxicity (Fig.6 B,C). Because the neuroprotective action of CZP against oxygen glucose deprivation model of cell death in organotypic hippocampal slice cultures does not result from an interaction with TRPV1 (Ray et al., 2003), additional experiments were performed to corroborate TRPV1-mediated neurotoxicity by using another TRPV1 antagonist, iodo-RTX (Rigoni et al., 2003), and showed similar results (data not shown).
40
Fig. 6. Effects of CZP (TRPV1 antagonist) on 6-OHDA (30 μM) or Spermidine (SPD) and Spermine (SPM)-induced neurotoxicity in dopaminergic neurons in neuron-enriched mesencephalic culture. The number of TH-ip neurons was counted in cultures treated with 6-OHDA (30 μM) (A), SPD (B), or SPM (C) for 24 hours in the absence or presence of CZP (TRPV1 antagonist). Graph represents the mean ± SEM of triplicate cultures for five separate platings expressed as a percentage of the control. *P < 0.05 compared with vehicle, #P < 0.05, ##P < 0.001 compared with 6-OHDA alone treated cultures (ANOVA and Student-Newman-Keuls analyses).