ABSTRACT
Chromosome 22q11.2 deletion syndrome is common and presents with multiple congenital anomalies ranging from congenital heart disease to cognitive and neuropsychiatric
conditions. Congenital large epigastric hernia is extremely rare in neonates. We present a case of a large congenital epigastric hernia in a neonate with DiGeorge syndrome.
Keywords: Abdominal hernia; 22q11.2 deletion syndrome; DiGeorge syndrome
INTRODUCTION
Epigastric hernia refers to protrusion of abdominal organs through openings in the linea alba, and occurs in 3%–5% of the population [1,2]. It is common in boys and is caused by tearing of the linea alba secondary to an increase in the intra-abdominal pressure. Epigastric hernias are usually small and can cause local pain [1]. Congenital epigastric hernia refers to herniation of intra-abdominal organs from the xiphoid process to the umbilicus due to failure of proper attachment of the rectus muscle during development [2]. DiGeorge syndrome is associated with cardiac deformities or maldevelopment of the thymus and thyroid glands; however, there are no reports of associated abdominal wall herniation [3].
Herein, we present an extremely rare case of a congenital large epigastric hernia that occurred in a neonate with DiGeorge syndrome and was successfully treated with surgery.
CASE REPORT
A 32-year-old healthy mother (gravida 0, para 0) delivered a female infant at 40 weeks and six days of gestation via emergency cesarean section due to complications of oligohydramnios.
The infant weighed 2.5 kilograms and Apgar scores were eight and nine at 1 and 5 minutes, respectively. The vital signs of the infant were stable on physical examination. A 4×3 cm abdominal mass was observed, covered in normal skin, which extended from the umbilicus to the xiphoid process. Intestinal movement was visible through the skin (Fig. 1).
The facial features of the baby were consistent with DiGeorge syndrome, including hypertelorism, a small chin, and deformed auricles (Fig. 2) [4]. Echocardiogram revealed a Received: Aug 26, 2020
Revised: Nov 22, 2020 Accepted: Dec 3, 2020 Correspondence to Jae Hee Chung
Department of Surgery, College of Medicine, The Catholic University of Korea, 222 Banpo- daero, Seocho-gu, Seoul 06591, Korea.
E-mail: [email protected] Copyright © 2021 Korean Association of Pediatric Surgeons
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (https://
creativecommons.org/licenses/by-nc/4.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ORCID iDs Hee Ju Kim
https://orcid.org/0000-0001-5438-5397 Jae Hee Chung
https://orcid.org/0000-0002-5866-119X Conflict of Interest
No potential conflict of interest relevant to this article was reported.
Author Contributions
Conceptualization: C.J.H.; Data curation:
K.H.J.; Writing - original draft: K.H.J., C.J.H.;
Writing - review & editing: C.J.H.
Hee Ju Kim , Jae Hee Chung
Department of Surgery, College of Medicine, The Catholic University of Korea, Seoul, Korea
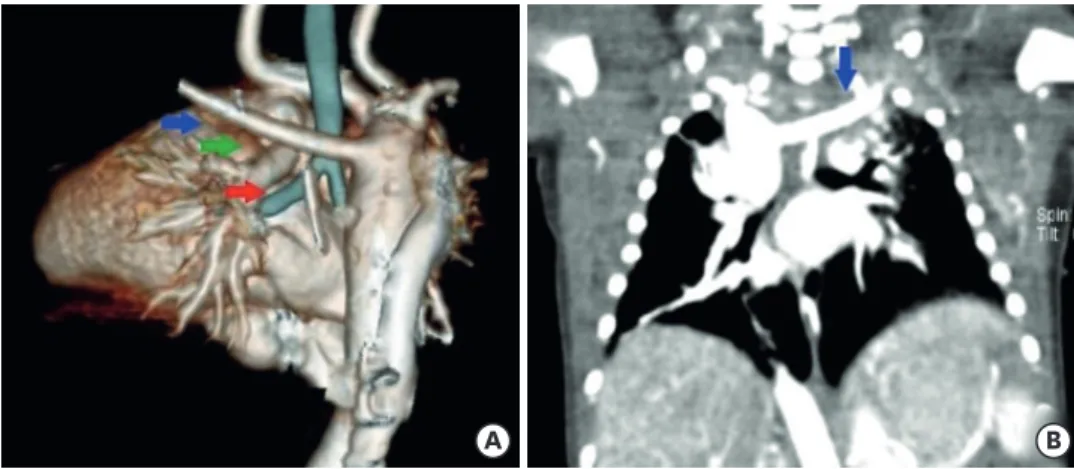
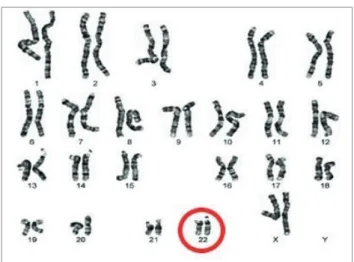
right aortic arch with an aberrant subclavian artery. Pulmonary 3D computed tomography angiography revealed an aberrant left subclavian artery, and the thymus was not observed (Fig. 3). Chromosome analysis revealed a microdeletion in chromosome 22, confirming the diagnosis of DiGeorge syndrome (Fig. 4). Abdominal ultrasonography and magnetic resonance imaging revealed the small intestine and left portion of the liver protruding through a 4.3×2.5×1.5 cm hernia sac. No musculoskeletal structure or linea alba was observed
A B
Fig. 1. Photographs of the congenital epigastric hernia.
A B
Fig. 2. Typical facial features characteristic of DiGeorge syndrome.
(A) Hypertelorism, narrowed palpebral fissures, prominent nose with anteverted nostrils, shortened philtrum, and a recessed and small chin and (B) malformed and low-set ears.
A B
Fig. 3. Pulmonary 3D CT angiography.
(A) Pulmonary 3D CT angiography showing right-sided aortic arch (green arrow), hypoplastic superior vena cava, prominent azygos vein (red arrow), and aberrant left subclavian artery (blue arrow) and (B) coronal view revealing aberrant left subclavian artery (blue arrow).
CT, computed tomography
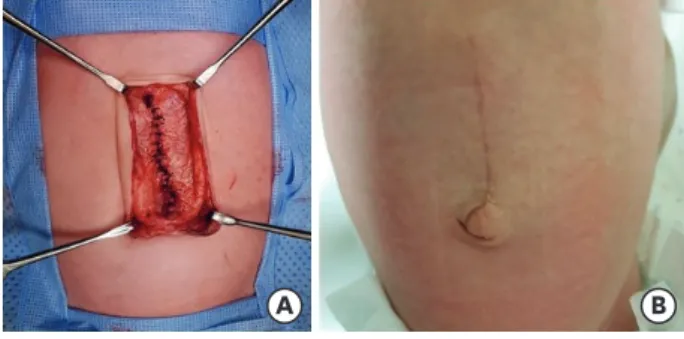
surrounding the hernia sac, and the rectus abdominis muscle was noted on either side of the sac (Fig. 5). The infant underwent successful repair of the congenital epigastric hernia from the xiphoid process to the umbilicus, on the fourth day of her life. There were no evident abnormalities or injury to the liver and small intestine during the surgery. After dissection of the peritoneum and fascia, we observed a fascial defect of approximately 3×4.5 cm. The baby recovered without complications after the surgery (Fig. 6) and was discharged on the 18th postoperative day.
DISCUSSION
In previous studies, congenital epigastric hernia has been characterized with various definitions due to the degree of abdominal wall protection., Furthermore, different names have been used to describe the condition [5,6]. Ciftci et al. [5] described the shape of a herniation as an “epigastric cleft,” wherein the rectus muscle had a normal shape, with the supraumbilical midline abdominal wall defect between the xiphoid process and the Fig. 4. Chromosome analysis.
Chromosomal study revealing 22q11.2 microdeletion.
A B
Fig. 5. Abdominal MRI.
MRI of the intestine showing a 4.3×2.5×1.5 cm hernia protruding in the anterior abdominal wall. It contains some portion of the small bowel and a part of the left liver. The subcutaneous fat is thin and abdominal wall muscle over the lesion is absent. There is no definite evidence of incarceration. (A) Axial view and (B) sagittal view.
MRI, magnetic resonance imaging.
umbilicus. Castañón et al. [6] described an abdominal wall defect in the midline, 2 cm below the xiphoid process, with evisceration of the greater omentum. They referred to the defect as congenital epigastric evisceration [6].
Epigastric hernia should be differentiated from diastasis recti. The chief difference between the two conditions is the presence of linea alba. Diastasis recti refers to the bulging of the weakened linea alba with an increase in the abdominal pressure, whereas epigastric hernia is a defect of the linea alba. Our case was distinct from diastasis recti due to the absence of the linea alba [7]. Therefore, we diagnosed this anomaly as a congenital epigastric hernia caused by abnormal formation of the abdominal wall due to failure of approximation of the linea alba.
Surgery should be planned in such cases for cosmetic purposes and to avert the possibility of intestinal incarceration or strangulation [8]. Although there were no immediate functional problems in this patient, the defect size was large, and a significant portion of the intestines and liver were dislocated. Prompt surgical repair was required to prevent improper
development of the abdominal cavity and the possibility of liver and intestinal damage from external injury.
DiGeorge syndrome, or 22q11.2 deletion syndrome, is a relatively common chromosomal microdeletion disorder that occurs in one in 2,000 to 4,000 individuals [3,8]. DiGeorge syndrome presents in a variety of clinical forms, with variable severity and timing of the symptoms. The classical abnormalities are immunodeficiency, hypoparathyroidism, congenital heart disease, dysmorphic facial features, hypocalcemia, and neurocognitive deficits [9]. The prognosis of DiGeorge syndrome varies depending on the severity of cardiac deformities; however, there is a 4%–14% overall risk of mortality [10]. Recently, the development of diagnostic methods has led to the survival of most patients to adulthood.
A study reported on connective tissue defects accompanying DiGeorge syndrome associated with congenital diaphragmatic hernia (CDH) [10]. CDH refers to the phenomenon in which the abdominal viscera herniates into the chest because of delayed closure of the diaphragm, and is associated with high mortality and morbidity [11]. The association between DiGeorge syndrome and CDH has been reported; however, abdominal wall defects have not been reported. Therefore, to the best of our knowledge, this study is the first report of a case wherein both conditions occurred simultaneously.
Nonetheless, there is no convincing evidence that epigastric hernia is a symptom of DiGeorge syndrome; therefore, the occurrence should be considered sporadic in our case. Additional
A B
Fig. 6. Closure of the hernia.
(A) The operative picture after closure of the muscle layer and (B) appearance on the seventh postoperative day.
approved the final manuscript, as submitted, and agree to be accountable for all aspects of the work. No funding was received for this study.
REFERENCES
1. Itani KMF. Umbilical and epigastric hernias. In: Evans SRT, ed. Surgical pitfalls: prevention and management. Philadelphia: W.B. Saunders; 2009. p.523-9.
2. Kelly KB, Ponsky TA. Pediatric abdominal wall defects. Surg Clin North Am 2013;93:1255-67.
PUBMED | CROSSREF
3. McDonald-McGinn DM, Sullivan KE, Marino B, Philip N, Swillen A, Vorstman JA, et al. 22q11.2 deletion syndrome. Nat Rev Dis Primers 2015;1:15071.
PUBMED | CROSSREF
4. Kilic SS, Gurpinar A, Yakut T, Egeli U, Dogruyol H. Esophageal atresia and tracheo-esophageal fistula in a patient with DiGeorge syndrome. J Pediatr Surg 2003;38:E21-3.
PUBMED | CROSSREF
5. Ciftci AO, Tanyel FC, Hiçsönmez A. Epigastric cleft: a previously unreported entity. J Pediatr Surg 1997;32:1247-9.
PUBMED | CROSSREF
6. Castañón M, Guimarães L, Tarrado X, San Vicente B, Albert A, Morales L. Congenital epigastric evisceration: a case report. J Pediatr Surg 2003;38:1253-4.
PUBMED | CROSSREF
7. Nahabedian MY. Management strategies for diastasis recti. Semin Plast Surg 2018;32:147-54.
PUBMED | CROSSREF
8. Coats RD, Helikson MA, Burd RS. Presentation and management of epigastric hernias in children. J Pediatr Surg 2000;35:1754-6.
PUBMED | CROSSREF
9. Rizvi S, Khan AM, Saeed H, Aribara AM, Carrington A, Griffiths A, et al. Schizophrenia in DiGeorge syndrome: a unique case report. Cureus 2018;10:e3142.
PUBMED | CROSSREF
10. Van L, Heung T, Graffi J, Ng E, Malecki S, Van Mil S, et al. All-cause mortality and survival in adults with 22q11.2 deletion syndrome. Genet Med 2019;21:2328-35.
PUBMED | CROSSREF
11. Unolt M, DiCairano L, Schlechtweg K, Barry J, Howell L, Kasperski S, et al. Congenital diaphragmatic hernia in 22q11.2 deletion syndrome. Am J Med Genet A 2017;173:135-42.
PUBMED | CROSSREF