Retroperitoneal Malignant Peripheral Nerve Sheath Tumors Complicated with Type I Neurofibromatosis
6
0
0
전체 글
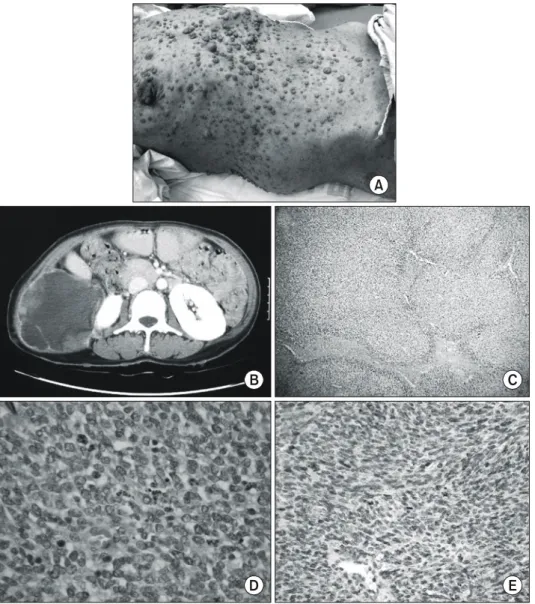
(2) 366. 대한외과학회지:제 71 권 제 5 호 2006. ꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏ. 검사실 소견: 입원 당시 혈액검사소견은 혈색소 10.9 g/dl, 헤마토크리트 32.9%, 백혈구수 17.2×103/ul, 혈소판 830× 3 10 /ul였으며, 일반화학 검사 소견은 모두 정상이었다. 방사선검사 소견: 복부 전산화 단층 촬영에서 우측 신장 주위에 장경 12 cm 가량의 불규칙한 경계를 가지고, 균일하 지 않은 low attenuation의 종괴가 관찰되었다. 종괴의 가장 자리는 조영증강의 소견을 보였다. 또한 이 종괴는 우측 복 부의 근육 일부를 침범하고 있었으며, 복강 내로 자라 들어 가는 양상을 보였다(Fig. 1). 수술 소견: 전신 마취 후에 종괴 위로 횡절개를 가했다. 종괴는 후복막의 우측 신장 외측으로 위치하면서 측후방으 로 복벽의 근육에 침윤이 의심되었다. 복강 내 원격전이의. 소견은 없었다. 복벽의 일부를 포함하여 광범위 절제를 시 행하였으나, 후벽에는 종괴의 일부가 남아 육안적 완전 절 제를 할 수 없었다. 절제한 종양의 최대장경은 11 cm였다 (Fig. 1). 남아있는 종괴의 주위로 클립을 이용하여 경계를 표시하고 수술을 마쳤다. 병리 소견: 검체는 11×11×9 cm 크기의 침윤성 종괴로, 단면에서는 현저한 출혈과 괴사를 보였다. 종괴는 다양한 핵형(pleomorphic nuclei)과 빈번한 세포분열을 보이는 방추 형 세포로 구성되었고, 지도 모양의 괴사(geographic necrosis)를 보였다. 단클론항체를 이용한 면역조직화학염색의 결과 Epithelial membrane antigen (EMA), Pan-cytokeratin (Panck)에 음성 반응을, Vimentin, CD56에 양성반응을 보였다.. A. B. C. D. E. Fig. 1. Clinicopathologic findings of case 1. (A) Patient have multiple neurofibromas of variable size and cafe-au-lait spots. (B) Abdominal CT scan present an ill-defined soft tissue mass in the right retroperitoneal space, which invades into peritoneal cavity and disrupts abdominal wall. (C) Microscopically tumor is composed of spindle cells and has multiple foci of geographic necrosis. (D) In high-power fields, mitosis is frequently observed and the tumor cells have pleomorphic nuclei, forming focally palisading pattern. (E) The tumor cells were negative for S-100 expression..
(3) 구본욱 외:제1형 신경섬유종증 환자에서 발생한 후복막의 악성말초신경초종양. 367. ꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏ. 최종 병리진단은 악성말초신경초종양이었다. 수술 후 경과: 환자는 수술 후 별 문제 없이 퇴원하였고, 방사선종양학과에서 6주에 걸쳐 5,940 cGy 용량으로 방사선 치료를 받았다. 술 후 6개월째 종괴가 있던 부위에 재발이 확 인되었고, 서혜부 림프절 종대도 관찰되었다. 더 이상의 적극 적인 치료를 원하지 않았으며, 술 후 10개월째 사망하였다. 증 례 2 환 자: 이○○, 27세 남자 주 소: 통증을 동반한 좌상복부 종괴 현병력: 환자는 내원 3개월 전부터 복부 팽만과 구토가 심해 식사를 제대로 하지 못해, 인근 의원에서 위내시경 검 사를 받고 소화성 궤양으로 치료받았다. 그러나 증상 호전 이 없고 체중 감소가 심하여, 본원 내과 외래에서 다시 위내 시경 검사를 시행하였다. 내시경 검사에서 위벽을 바깥에 서 누르는 소견을 보이고, 신체 검사에서 좌상복부에 종괴 가 촉지되어 입원하였다. 복부 팽만은 좌상복부의 종괴에 의한 것으로 생각되었고, 경과 관찰 기간 동안 눈에 띄게 커졌다. 환자는 어려서부터 지능 저하가 있었으며 전신에 다수의 카페오레 반점과 다양한 크기의 피부 결절이 있었. 으나, 신경섬유종증 진단을 받지 않고 지내왔다. 과거력 및 가족력: 군복무 시절 고혈압을 진단받았는데, 환자의 아버지와 할머니도 고혈압으로 치료를 받고 있다. 환자는 간헐적으로 혈압 강하제를 경구 복용하는 외에는 특별한 치료를 받은 적이 없었다. 신경섬유종증의 가족력 은 없었다. 신체검사 소견: 환자의 매우 쇠약해 있었고, 계속되는 통 증으로 지쳐 있었다. 내원 전 3개월 동안 5 kg의 체중 감소 (증상발현 전 54 kg/170 cm)가 있었다. 입원 당시 환자의 혈 압은 140/90 mmHg, 맥박수 126회/분, 호흡수 25회/분, 체온 o 37.8 C였다. 전신에 다수의 카페오레 반점과 다양한 크기의 신경섬유종이 관찰되었다. 종괴는 좌상복부 늑골하연 아래 로 최대 장경 20 cm 정도의 거대 종괴가 촉지되었다. 종괴 는 고정된 느낌이었고, 압통이 있었다. 그 외 복부 진찰 소견 은 정상이었다. 신경학적 검사에서도 특이 소견은 없었다. 검사실 소견: 입원 당시 혈액검사소견은 혈색소 7.0 g/dl, 3 헤마토크리트 22.2%, 백혈구수 9.7×10 /ul, 혈소판 405× 3 10 /ul였으며, 일반화학 검사 결과 이상 소견은 Calcium 8.0 (참고치 8.2∼10.8) mg/dl, Phosphours 2.1 (2.5∼5.5) mg/dl, Glucose 148 (70∼110) mg/dl, Cholesterol 73 (130∼230). A. B. C. D. Fig. 2. Clinicopathologic findings of case 2. (A) Abdominal CT scan present a heterogenously enhancing soft tissue mass, which invades into posterior abdominal wall. (B) A huge retroperitoneal soft tissue mass displaces stomach anteriomedially and spleen and pancreas superiorly. (C) Cut surface of the resected mass shows marked hemorrahge and necrosis. (D) Microscopically tumor is composed of spindle cells with pleomorphic nuclei and frequent mitosis..
(4) 368. 대한외과학회지:제 71 권 제 5 호 2006. ꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏ. mg/dl, Protein 5.8 (6.5∼8.2) g/dl, Albumin 2.2 (3.5∼5.1) g/dl, AST (SGOT) 147 (0∼40) IU/L, ALT (SGPT) 83 (0∼40) IU/L, Alk. Phosphatase 937 (65∼220) IU/L이었다. P.T (%)는 67.9 (80∼100)%였고, a PTT는 46.7 (25.0∼35.5)초로 연장되 어 있었다. 부신의 기능성 종양 가능성을 배제하기 위해 시 행한 검사는 모두 정상 범위였다. 방사선검사 소견: 복부 전산화 단층 촬영에서 좌상복부 에 불규칙한 조영 증강을 보이는 거대 종괴가 관찰되는데, 좌측 부신이나 그 주위의 후복막에서 기원한 것으로 보였 다. 비장과 위는 종괴에 의해 밀려 있고, 복강 내 소량의 복 수가 관찰되었다(Fig. 2). 전신 골주사 검사에서는 전체 골 격에 미만성 증가 소견을 보여 대사성 골진환을 의심하는 소견이었다. 골밀도 검사는 정상 소견이었다. 수술 소견: 전신 마취 후에 정중선 절개를 시행하였다. 복강 내 소량의 복수가 관찰되었으나, 원격전이의 소견은 없었다. 종괴는 후복막의 좌측 신장 위쪽에 위치하면서 대 동맥, 비동맥 및 비정맥과 단단히 유착되어 분리할 수 없었 다. 대동맥 벽에 종양의 일부를 남기고, 비장과 췌미부를 포 함하여 종양을 절제하였다. 절제한 종양의 최대장경은 15 cm였다(Fig. 2). 절제 후 대동맥 주위에 남긴 종양에서 출혈 이 계속되어 거즈로 누르고 수술을 마쳤다. 수술 후 48시간 에 다시 개복하여 거즈를 제거하고 출혈이 없음을 확인한 뒤 배액관을 삽입하고 수술을 마쳤다. 병리 소견: 검체는 15×10×8 cm 크기의 종괴로, 단면에 서는 현저한 출혈과 괴사를 보였다. 종괴는 다양한 핵형 (pleomorphic nuclei)과 빈번한 세포분열을 보이는 방추형 세 포로 구성되어 있었다. 단클론항체를 이용한 면역조직화학 염색의 결과 SMA, CK, S-100, HMB-45에 음성 반응을, Vimentin에 양성반응을 보였다. 최종 병리진단은 악성말초 신경초종양이었다. 비장과 췌장에 종양의 침윤은 없었다. 수술 후 경과: 환자는 술 후 3주째 퇴원하였는데, 복부팽 만은 소실되고, 통증은 감소하였지만, 식사를 제대로 하지 못하고 체중은 계속 감소하였다. 방사선 치료를 계획하였 으나, 환자가 거부하였다. 술 후 1개월에 시행한 초음파 검 사에서 대동맥 주위에 남긴 종괴가 장경 6 cm 크기로 커진 것을 관찰하였다. 환자는 술 후 3개월에 사망하였다.. 고. 찰. 신경섬유종증은 신경계와 피부 병변이 주를 이루지만, 뼈, 연부조직을 포함하여 거의 모든 장기를 침범하는 체세 포 우성 유전의 유전질환이다. 여러 가지 아형이 발표되었 는데, 제1형 신경섬유종증이 가장 흔하며, 세계적으로 3,500 명의 신생아에서 1명꼴로 발생한다.(3-5) 제1형 신경섬유종 증은 종양 발생 가능성이 매우 높은 유전성 질환인데, 신경 섬유종이 가장 흔히 동반되며, 피부형(cutaneous), 피하형 (subcutaneous), 망상(plexiform)의 세 유형으로 나눌 수 있다.. 신경섬유종은 여러 세포로 구성되지만, Schwann 세포가 주 를 이루며, 이 세포가 특징적인 유전적 변이를 보이는 종양 의 전구세포(progenitor)이다.(6,7) 악성말초신경초종양(malignant peripheral nerve sheath tumor)은 제1형 신경섬유종증에 서 발생할 수 있는 악성종양으로 사망의 주원인 중 하나이 다. 새롭게(de novo) 발생할 수도 있지만, 대부분 섬유종 특 히 망상 섬유종에서 유래한다.(8,9) 악성의 진단은 10HPF에 서 5개 이상의 mitotic count를 보일 때, 세포의 이형성이 증 가한 경우, 종양에 괴사가 있을 때, 종양의 경계가 침윤성일 때, 주변 조직에 침습이 있는 경우 등에서 내릴 수 있다. 제1 형 신경섬유종증 환자에서 섬유종이 커지고 통증을 유발하 는 경우에는 반드시 절제 또는 조직검사를 통해 악성말초 신경초종양의 가능성을 확인해야 한다. 본 증례들의 경우 에도 종괴 부위의 통증과 압통이 환자들이 병원을 찾은 주 원인이었다. 악성말초신경초종양은 드문 신경외배엽 기원의 종양으 로 neurofibrosarcoma, malignant neurilemmoma, malignant schwannoma, neurogenic sarcoma 등 여러 이름으로 불린다. 연부육종의 5% 정도를 차지하는데, 이 중 절반 정도가 제1 형 신경섬유종증 환자에서 발생한다. 제1형 신경섬유종증 환자에서 일생동안 악성말초신경초종양이 발생할 위험은 2∼13% 정도이다.(1,2) 발암의 기전은 제1형 신경섬유종증 에 나타나는 chromosome 17q11.2의 NF1 유전자 돌연변이 에, 추가로 p53이나 CDKN2A 등의 유전자 변이가 동반될 때 악성말초신경초종양이 발생하는 것으로 추정하고 있 다.(10-12) 더불어 방사선 조사는 잘 알려진 발암 위험 요인 으로, 평균 잠복기(latency period)는 15년 정도이다.(13,14) 본 증례들의 경우 가족력이 없는 속발성 제1형 신경섬유종 증 환자들로서 방사선 조사의 과거력은 없었다. 악성말초신경초종양은 남녀 모두에서 비슷한 빈도로 생 긴다.(15,16) 제1형 신경섬유종증 환자에서의 발생 연령은 일반인보다 빨라서 평균 30세 정도이다.(15) 사지가 가장 흔히 발생하는 부위로 전체의 50% 이상을 차지하며, 다음 으로 두경부, 체간의 순으로 호발한다. 후복막은 비교적 환 자수가 많은 보고에서도 10년간 1∼2예 정도로 나타나며, 전체 악성말초신경초종양의 5∼8%를 차지한다.(15-18) 악 성말초신경초종양은 국내에서도 드물게 증례보고 되었으 나, 본 증례와 같이 제1형 신경섬유종증 환자에서 후복막에 발생한 경우는 매우 드물다.(19,20) 악성말초신경초종양은 가장 공격적인 암의 하나로 심부 에 위치하거나 인접한 주요 장기의 국소침윤으로 외과적 완전절제가 어려운 경우가 많다. 본 증례들의 경우에도 종 양이 복벽을 광범위하게 침범하거나, 복부 대동맥의 상당 부분을 침범하여 완전절제가 불가능하였다. 문헌상 육안적 으로 완전 절제가 가능한 경우는 전체의 38∼69% 정도인 데, 종양의 위치에 따라 달라질 수 있다.(15,16) 즉, 척추옆 (paraspinal)에 있는 경우는 20% 미만이지만, 사지에 있는 경.
(5) 구본욱 외:제1형 신경섬유종증 환자에서 발생한 후복막의 악성말초신경초종양. 369. ꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏ. 우에는 95% 이상에서 완전 절제가 가능하다. 종양 절제 시 약 절반에서 원발 신경을 찾을 수 있다고 하나 본 증례들에 서는 찾을 수 없었다.(15,16) 후복막의 척추옆에 발생한 악성말초신경초종양의 수술 시 특히 출혈에 유의해야 하는데, 대동맥 분지가 많고, 혈관 들이 매우 약해, 결찰하거나 봉합하기 어렵기 때문이 다.(17,21) 증례 2의 경우 수술 중 지혈에 어려움이 있어 거 즈로 누르고 48시간 후에 이를 제거하였다. 종양의 절제 전 에 충분하게 주요 혈관의 근위부와 원위부를 확보해 출혈 에 대비하는 것이 중요하겠다. 육안적인 완전 절제 후에도 평균 12∼15개월 뒤 24∼38%에서 국소적으로 재발되며, 절 제연이 육안적으로도 불충분한 경우에는 훨씬 빨리 재발한 다.(15,16,18) 대부분의 악성말초신경초종양은 악성도가 높은(high-grade) 종양으로(low-grade는 10∼15%) 국소재발이나 원격전이의 빈도가 높다.(1) 외과적 절제 후 평균 16∼22개월 뒤 20∼ 52%에서 원격전이를 진단하게 된다.(15,16) 악성말초신경 초종양은 수술 후 재발이 빈번한 데도 불구하고 효과적인 보조 항암 요법이 없다. 기존의 항암 화학요법은 반응성 매 우 낮아 치료 효과를 기대하기 어렵다.(16,17) 일부 후향적 분석에서 술 후 방사선 조사가 국소 재발을 줄여준다고 하 지만, 효과가 없다는 보고도 있다.(22) 본 증례들 중 1예에서 술 후 방사선 조사가 시행되었지 만, 종양의 성장을 억제하지 못했다. 악성말초신경초종양의 전체 5년 생존율은 30∼50% 정도이다.(1,15,18) 잘 알려진 예후인자로는 종괴크기(5 cm 또는 10 cm), 조직학적 악성도 (grade), 육안적 완전 절제(gross total resection) 등이 있 다.(15,16,22) 발생 연령, 발생부위, 제1형 신경섬유종증 환 자인지 여부가 예후에 유의한 영향을 미치는지는 보고자에 따라 견해가 다르다. 발생 부위의 경우, 기본적으로 완전 절 제의 가능성에 차이를 보이겠지만, 예후와 직접 연관은 없 다는 보고도 많다.(15-17) 일반적으로 환자의 성별, 절제연 의 조직학적 소견(positive margin), 술 후 보조 화학요법 등 은 예후와 무관하다.(15,16, 22) 본 증례들의 경우 술 후 생 존 기간이 매우 짧은 데, 우선 육안적 완전절제가 불가능한 경우였다는 점이 큰 영향을 미친것으로 생각한다. 증례 2의 경우에는 수술 전 환자의 전신 상태를 개선해 보려고 노력 하였으나, 이 기간 동안에도 현저하게 종양이 커지고, 정맥 영양 등의 보조적 치료에도 전신 상태가 개선되지 않아 서 둘러 수술을 시행하였으나, 환자의 생존을 유의하게 연장 하지는 못하였다. 현재 악성말초신경초종양의 치료는 외과적 절제에 전적으 로 의존하고 있는 만큼, 제1형 신경섬유종증 환자에서 주기 적인 진찰과 검사를 통해 악성말초신경초종을 조기에 발견 하여 절제하는 것이 환자의 예후를 개선하는 데 가장 중요하 겠다. 더불어 새로운 효과적인 치료법의 개발이 필요하겠다.. REFERENCES 1) Woodruff JM. Pathology of tumors of the peripheral nerve sheath in type 1 neurofibromatosis. Am J Med Genet 1999; 89:23-30. 2) Evans DG, Baser ME, McGaughran J, Sharif S, Howard E, Moran A. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J Med Genet 2002;39:311-4. 3) Huson SM. Recent developments in the diagnosis and management of neurofibromatosis. Arch Dis Child 1989;64:745-9. 4) Rasmussen SA, Friedman JM. NF1 gene and neurofibromatosis 1. Am J Epidemiol 2000;151:33-40. 5) Poyhonen M, Kytola S, Leisti J. Epidemiology of neurofibromatosis type 1 (NF1) in northern Finland. J Med Genet 2000; 37:632-6. 6) Peltonen J, Jaakola S, Lebwohl M. Cellular differentiation and expression of matrix genes in type 1 neurofibromatosis. Lab Invest 1988;59:760-71. 7) Frahm S, Mautner VF, Brems H, Legius E, Debiec-Rychter M, Friedrich RE, et al. Genetic and phenotypic characterization of tumor cells derived from malignant peripheral nerve sheath tumors of neurofibromatosis type 1 patients. Neurobiol Dis 2004;16:85-91. 8) Gutmann DH, Collins FS. Neurofibromatosis type 1. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill; 1995. p. 76-96. 9) Scheithauer BW, Woodruff JM, Erlandson RA. Neurofibroma, tumors of the peripheral nervous system. In: Scheithauer BW, Woodruff JM, Erlandson RA, editors. Atlas of Tumor Pathology, Third Series. Washington, DC: Armed Forces Institute of Pathology; 1999. p. 177-218. 10) Gutmann DH, Wood DL, Collins FS. Identification of the neurofibromatosis type 1 gene product. Proc Natl Acad Sci 1991;88:9658-62. 11) Berner JM, Sorlie T, Mertens F, Henriksen J, Sater G, Mandahl N, et al. Chromosome band 9p21 is frequently altered in malignant peripheral nerve sheath tumors: studies of CDKN2A and other genes of the pRB pathway. Genes Chromosomes Cancer 1999;26:151-60. 12) Birindelli S, Perrone F, Oggionni M, Lavarino C, Pasini B, Vergani B, et al. Rb and TP53 pathway alterations in sporadic and NF1-related malignant peripheral nerve sheath tumors. Lab Invest 2001;81:833-44. 13) DeCou JM, Rao BN, Parham DM, Lobe TE, Bowman L, Pappo AS, et al. Malignant peripheral nerve sheath tumors: The St Jude Children’s Research Hospital experience. Ann Surg Oncol 1995;2:524-9. 14) Ducatman BS, Scheithauer BW. Postirradiation neurofibrosar.
(6) 370. 대한외과학회지:제 71 권 제 5 호 2006. ꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏ coma. Cancer 1983;51:1028-33. 15) Ramanathan RC, Thomas JM. Malignant peripheral nerve sheath tumours associated with von Recklinghausen's neurofibromatosis. Eur J Surg Oncol 1999;25:190-3. 16) Baehring JM, Betensky RA, Batchelor TT. Malignant peripheral nerve sheath tumor: the clinical spectrum and outcome of treatment. Neurology 2003;61:696-8.. 17) Neville H, Corpron C, Blakely ML, Andrassy R. Pediatric neurofibrosarcoma. J Pediatr Surg 2003;38:343-6. 18) Lee JS, Jeon DG, Cho WH, Lee SY, Oh JM, Kim JW. Treatment and survival rate of malignant peripheral nerve sheath tumors. J Korean Bone Joint Tumor Soc 2003;9:131-8. 19) Seon IC, Han CH, Kwak KM, Chung WK, Kang SH, Sin OL.. Retroperitoneal malignant schwannoma. Korean J Urology 2002;43:250-2. 20) Choi KS, Whang YN, Kim YJ, Yang YM, Cho SW, Hong YK, et al. A case of soliatry malignant schwannoma in the retroperitoneum. Korean J Med 1988;35:736-827. 21) Nah YW, Suh JH, Choi DH, Ko BK, Nam CW, Kim GY, et al. Benign retroperitoneal schwannoma: surgical consideration Hepatogastroenterology 2005;52:1681-4. 22) Carli M, Ferrari A, Mattke A, Zanetti I, Casanova M, Bisogno G, et al. Pediatric malignant peripheral nerve sheath tumor: the Italian and German soft tissue sarcoma cooperative group. J Clin Oncol 2005;23:8422-30..
(7)
수치

관련 문서
Karyotype results showed a mosa- ic loss in the Y chromosome of the malignant cells, but not in the normal and benign cells, suggesting that the somatic loss of the Y chromosome in
