대한소화기학회지 2005;45:78-87 □REVIEW □
ꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏ 접수: 2004년 8월 1일
연락처: 박재갑, 110-744, 서울특별시 종로구 연건동 28번지 서울대학교 의과대학 암연구소
Tel: (02) 2072-3380, Fax: (02) 742-4727 E-mail: [email protected]
ꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏ Correspondence to: Jae-Gahb Park, M.D., Ph.D.
Korean Polyposis Registry, Laboratory of Cell Biology Cancer Research Insititute and Cancer Research Center Seoul National University College of Medicine 28 Yongon-dong, Chongno-gu, Seoul 110-744, Korea Tel: +82-2-2072-3380, Fax: +82-2-742-4727 E-mail: [email protected]
유전 대장암
서울대학교 의과대학 암연구소
박재갑․김일진
Hereditary Colorectal Cancer
Jae-Gahb Park, M.D., Ph.D., and Il-Jin Kim, Ph.D.
Research Institute and Cancer Research Center, Seoul National University College of Medicine, Seoul, Korea
Hereditary syndromes cause approximately 5 to 15% of overall colorectal cancer (CRC) cases. Hereditary CRC is conventionally divided into two major categories: hereditary non-polyposis colorectal cancer (HNPCC) and those related to polyposis syndromes including familial adenomatous polyposis (FAP), Peutz-Jegher syndrome (PJS), and juvenile polyposis (JP). The screening for the cancer and methods of treatment applied to patients with hereditary CRC are quite different from those applied to the general population. The genes responsible for these syndromes has recently identified, as a result, genetic testing has become the most important determining factor in clinical decisions. Germ-line mutation of the APC gene induces FAP, an autosomal dominant disorder, characterized by the development of hundreds to thousands of colonic adenomas. CRC appears in almost all affected individuals by the time they are 50 years of age. An affected individual should undergo colectomy by his/her late teens.
Furthermore, according to the findings of genetic testing, at-risk family members also need endoscopic sur- veillance and surgery. Recently, a mutation on the MYH gene is increasingly being investigated in patients with multiple polyps, and autosomal recessive MYH polyposis is considered to be a new category of polyposis. More common than FAP, HNPCC is caused by germ-line mutations in DNA mismatch repair genes, mainly MLH1 and MSH2. Although there is no polyposis, polyps seem to be more villous and dysplastic and appear to grow rapidly into CRCs. The aggregate lifetime risk of CRC is about 80% for mutation carriers. The risk for other types of cancer, such as endometrial, ovarian, small bowel, and transitional cell cancer, is also increased. The Amsterdam criteria and Bethesda guidelines are the best-known tools for diagnosis and genetic testing, and colectomy followed by endoscopic follow-up is the standard treatment. PJS and JP are reported to be characterized by hamartomatous polyps throughout the GI tract and germ-line mutations in the STK11 gene (PJS) and the DPC4/BMPR1A gene (JP). (Korean J Gastroenterol 2005;45:78-87)
ꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏꠏ
Key Words: Hereditary colorectal cancer; Familial adenomatous polyposis; Hereditary non-polyposis colorectal
cancer; Peutz-Jegher syndrome; Juvenile polyposis
박재갑 외 1인. 유전 대장암 79
서 론
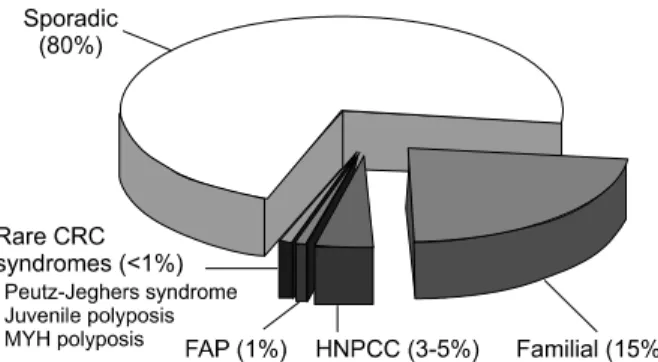
대장암은 우리나라에서 전체 암 중 제 4위의 발생빈도1를 나타내는 비교적 흔한 암일 뿐만 아니라 지속적인 증가 추 세를 보이고 있다. 대장암의 원인으로는 환경 요인, 특히 식 이 섭취 양상이 주로 관여하며, 전체 대장암 환자의 약 20%
는 유전 요인에 의해 발생한다(Fig. 1).2
유전 요인에 의해 발생하는 대장암은 환경 요인에 의해 발생하는 경우와는 달리 원인이 명확한 경우가 많다. 또한 출생 시부터 결함이 있는 유전자를 갖고 태어나므로 일반인 에서보다 대장암의 발생이 일찍 나타나며, 유전자의 기능 이상이 대장에만 국한된 것이 아니기 때문에 대장 외 장기 에도 이상 소견을 나타내는 경우가 많다.3
유전 성향을 나타내는 질환으로서 대장암과 관련 있는 대 표적인 것으로는 유전 대장용종증 증후군이 있다. 이는 대 장에서 다발성으로 용종이 생기는 질환들을 총칭하는 것으 로서, 여기에 속하는 질환으로는 연소기 용종증, 포이츠-예 거스 증후군, 코우덴 증후군, 토레 증후군, 터코트 증후군 그리고 유전 대장용종증 증후군의 가장 대표적인 질환인 가 족 용종증 등이 있다. 가족 용종증과 같이 선종이 다발성으
로 생기는 질환들은 각각의 선종이 대장암으로 진행될 가능 성이 특별히 더 높지는 않지만 수백, 수천 개의 선종이 존재 하기 때문에 대장암이 발생할 가능성은 매우 증가한다. 실 제로 가족 용종증의 경우는 치료하지 않으면 100%에서 대 장암으로 진행한다.4 포이츠-예거스 증후군과 연소기 용종 증 등 주로 과오종 용종증을 나타내는 경우에는 이러한 과 오종 암의 전구병변은 아니지만 이 병을 가진 환자들의 경 우 정상인보다 대장암이 발생할 위험이 훨씬 크다는 점에서 유전 대장암의 한 범주로 취급하고 있다.
유전 대장암에 속하는 또 다른 대표적인 질환으로는 멘델 우성 유전을 하는 것이 밝혀진 유전 비용종증 대장암이 있 다. 이 질환은 현재까지 알려진 유전 종양 중 가장 흔한 발 생빈도를 보이므로 임상적으로 매우 중요하다.
유전 대장암을 일으키는 질환 유전자는 이미 알려져 있거 나 현재 연구 중인데, Table 1에 원인 유전자가 이미 밝혀진 질환의 원인 유전자를 나타내었으며 Table 2에 각 질환들의 특성을 나타내었다.
최근에는 멘델 열성 유전을 하면서 대장 내에 다발 선종 과 암이 발생하는 질환으로 염기절제교정(base excision re- pair)유전자 중 하나인 MYH 유전자의 배선돌연변이에 의한 MYH 용종증(MYH-associated polyposis, MAP)이 보고되었 다.5
본 론
1. 가족 용종증
가족 용종증은 유전 대장용종증 증후군의 가장 대표적인 것으로서 선종들이 전 대장에 걸쳐 100개 이상 산재되어 나 타나는 질환으로 조기에 예방 대장절제술을 시행하지 않으 면 100%에서 악성화하여 대장암으로 진행한다. 이 질환은 멘델 우성 유전을 하며, 환자의 70-80%에서는 가족 용종증 의 가족력이 있지만 약 20-30%에서는 가족력이 없이 당대 Fig. 1. Frequency of hereditary colorectal cancer.
CRC, colorectal cancer; FAP, familial adenomatous polyposis;
HNPCC, hereditary nonpolyposis colorectal cancer.
Table 1. Genetics of Inherited Colorectal Tumor Syndromes
Syndrome Gene Location
Familial adenomatous polyposis (FAP) APC 5q Hereditary nonpolyposis colorectal cancer
(HNPCC)
MSH2, MLH1, PMS1, PMS2, MSH6, TGF-βRII
2p, 3p, 2q, 7p 2p, 3p
Peutz-Jeghers syndrome STK11 (LKB1) 19p
Juvenile polyposis syndrome SMAD4 (DPC4) BMPR1A
18q 10q22.3
MYH polyposis MYH 1p
APC, adenomatous polyposis coli; STK, serine threonine kinase; SMAD, Sma and Mad related protein; BMPR1A, bone morphogenetic protein receptor type 1A.
80 The Korean Journal of Gastroenterology: Vol. 45, No. 2, 2005
의 돌연변이 형태로서 나타난다.6 가족 용종증에 동반되어 발생하는 대장암은 전체 대장암 환자의 1% 정도를 차지한 다. 이 질환은 근본적으로 모든 세포 성장과정의 조화가 와 해된 질환이므로 대장 이외에도 여러 장기에 다양한 병변을 동반할 수 있는 질환이다.
1) 가족 용종증의 임상상
가족 용종증에 이환된 환자는 주로 사춘기를 전후하여 용 종이 발생하는데 대장에 선종이 생기는 연령의 중앙치는 16 세(범위는 5-38세 사이)이고, 대장암의 발생률은 20세에 0.5%, 25세에 4%, 30세에 13%, 35세 때에는 23%, 40세에서 는 37%나 되며 그 이후로도 연령에 따라 계속적으로 증가 한다. 치료하지 않은 가족 용종증 환자의 자연경과를 보면, 선종의 발생은 평균 25세, 증상 발현은 33세, 선종의 진단은 36세, 대장암의 진단은 39세이고 평균 42세에 대장암으로 사망한다.7
가족 용종증에서 나타날 수 있는 대장 이외의 병변은 아 주 다양하다. 동반되는 양성 병변으로 안면과 긴 관상골에 주로 발생하는 골종양, 피지낭포증, 치아의 이상, 눈에 나타 나는 망막색소상피의 선천 비대(congenital hypertrophy of the retinal pigment epithelium, CHRPE) 등이 있다. 가족 용종증 에 동반되어 나타나는 종양 병변으로는 간모세포종, 유건종 (desmoid), 갑상선암, 십이지장에 발생하는 선종 및 선암, 소 장의 용종 및 암, 위의 용종 및 암, 담도계 및 췌장의 선종, 뇌종양 등이 있다.8 사망 원인은 대장암이 대부분의 원인이었 으나 대장암이 발생하기 이전에 예방 대장절제술을 시행하는 것이 보편화된 이후에는 유건종, 십이지장암, 갑상선암 및 위 암 등 대장 이외의 병변이 주요한 원인이 되고 있다.
가족 용종증에 대한 치료는 대장절제술이다. 대장암 발생 의 위험 때문에 25세 이전에는 반드시 수술을 해주는 것이 원칙이다. 증상이 없는 5 mm 이하의 용종을 가진 환자들에 서는 증상이 발현되거나 20세의 연령이 되면 수술을 해 주 고, 증상이 있는 환자들이나 5-10 mm 이상의 용종을 가지거 나 조직검사 소견에서 이형성증이 발견되면 즉시 수술을 시 행하여야 한다. 수술 방법은 여러 가지가 있지만 전 결장과 상부직장 절제 및 하부직장 점막 절제 후 회장저장낭을 만 들어 항문에 문합해 주는 방법이 선호된다. 용종의 발생 및 성장을 억제하기 위한 수단으로 비스테로이드 소염진통제 인 설린닥이나 선택 COX-2 (cyclooxygenase-2) 억제제인 셀 레콕시브 등을 경구 투여하는 경우 용종의 크기 및 수가 감 소하지만, 용종을 완전히 없애지는 못한다.9,10
2) 가족 용종증의 발생 원인 (1) APC 유전자
1987년 보드머 등에 의해서 가족 용종증에 관여하는 유전 자가 5번 염색체의 장완(5q 21-22부위)에 위치하는 것이 밝 혀졌으며,11 1991년 이 부위에서 발견된 몇 개의 종양억제유 전자 중에서 가족 용종증의 원인이 되는 유전자인 APC 유 전자가 밝혀진 바 있다.12 APC 유전자는 15개의 엑손으로 구성되어 있으며 총 2,843개의 아미노산으로 이루어진 단백 질을 생성하는데 이 단백질은 세포들 사이의 부착에 관여하 는 단백질인 베타카테닌이라는 단백질과 결합하여 세포 사 이의 신호전달에 관여함으로써 세포의 성장 조절에 영향을 미친다. 가족 용종증은 APC 유전자의 배선돌연변이에 의해 생성된 비정상 단백질에 의해 발생한다. 현재까지 밝혀진 바에 의하면 APC 유전자의 돌연변이는 이 유전자의 5’쪽 Table 2. Hereditary Colorectal Cancers*
HNPCC FAP Peutz-Jeghers
syndrome Juvenile polyposis MYH polyposis Pattern Autosomal dominant Autosomal dominant Autosomal dominant Autosomal dominant Autosomal recessive Causative genes hMLH1/hMSH2/hPMS1
hPMS2/hMSH6/hMLH3 APC STK11 DPC4/
BMPR1A MYH
Frequency 5% 1% 0.1% 0.1% Unknown
Organ involved Colon Colon Small bowel Colon Colon
Frequency of polyp 20-40% 100% >90% >90%
Number of polyp 1-10 >1,000 10-100 50-200 3-100
Malignancy risk 80% 100% 50% 10-20%
Age occurring
malignancy 5th decade 25 4th decade 4th decade 6th decade
* Previously presented in The 9th Congress of the Asian Federation of Coloproctology (AFCP) 2003.
HNPCC, hereditary nonpolyposis colorectal cancer; FAP, familial adenomatous polyposis; APC, adenomatous polyposis coli; STK, serine threonine kinase; SMAD, Sma and Mad related protein; BMPR1A, bone morphogenetic protein receptor type 1A.
Park JG, et al. Hereditary Colorectal Cancer 81
1/2에 대부분(97%)이 위치하며 특히 엑손 15번의 코돈 1,000-1,600 사이에 60%가 위치하며, 전형적인 돌연변이는 염기쌍의 결손이지만 점돌연변이도 간혹 발견된다. 특징적 으로 약 95%에서 돌연변이에 의해 APC 단백질의 길이가 짧아진다.
(2) 발암 기전
APC 유전자는 종양억제유전자의 일종이므로 쌍으로 된 유전자 중 1개에서만 돌연변이가 발생해도 세포를 형질변 환시키는 암유전자와는 달리 2개에서 모두 돌연변이가 발 생하여 정상 역할을 하는 단백의 생성이 되지 않아야 암을 일으킨다. 가족 용종증의 발암 기전에서 APC 유전자의 역 할이 일반 대장암에서와 다르지는 않다. 다만 가족 용종증 에서는 출생 시부터 쌍으로 된 대립유전자 중 한 개가 이미 결손된 형태이므로 나머지 한 개에서 돌연변이로 결손될 가 능성이 일반인보다 훨씬 높을 뿐이다. APC 유전자는 다양 한 역할을 하는 유전자이기 때문에 아직까지 그 정확한 역 할은 알려져 있지 않지만 초파리의 실험으로부터 밝혀진 베 타카테닌 경로가 가장 잘 알려져 있다. 정상 APC 단백질이 생성되지 않으면 세포 내 베타카테닌의 분해가 이루어지지 않아 카테닌이 세포 내에 축적되어 세포 내 신호전달과정을 교란시킨다(Fig. 2).
3) 가족 용종증의 증상 발현 전 조기 진단
증상 발현 전 조기 진단이 가능하게 된 것은 가족 용종증 의 원인 유전자인 APC 유전자가 밝혀진 이후이다. 현재 증 상 발현 전 조기 진단에 사용되는 방법으로는 연관분석에 의한 진단, 염기서열 결정을 통한 진단, 단백질 검사를 통한 진단 등이 있다. 연관분석은 APC 유전자에 밀접하게 연관 되어 있으면서 높은 다형성을 나타내는 (CA)n 반복 구조를 증폭하여 증폭된 반복서열 길이가 다른 정도를 분석하면 가
족 용종증 환자의 가계 구성원 중에서 이상이 있는 APC 유 전자를 가지고 있는 구성원을 판별하는 것이다. 그러나 연 관분석에 의한 진단은 최소한 2세대에 걸쳐 각 세대마다 1 명 이상의 환자가 있는 가계에서만 적용 가능한 한계가 있 다.13 염기서열 결정을 통한 진단은 연관분석을 통한 유전자 진단이 불가능한 경우에 이용될 수 있는 방법으로서 우선 가족 용종증 환자 및 그 가족 구성원들로부터 채혈을 하여 백혈구에서 DNA를 분리한 뒤 중합효소연쇄반응을 이용하 여 유전자를 증폭시킨다. 증폭된 유전자를 단일쇄형태구조 다형성분석(single-strand conformation polymorphism)이나 리 보핵산분해효소 보호검색(ribonuclease protection assay)을 시 행하여 APC 유전자에서 돌연변이가 존재하는 부위를 먼저 찾고 그 부위의 염기서열을 봄으로써 돌연변이를 확인하는 방법이다. APC 유전자는 지금까지 밝혀진 유전자 중 가장 큰 것 중의 하나여서 전체 유전자의 염기서열을 결정하는 것은 많은 시간이 소요되므로 이와 같은 방법을 사용한다.
APC 유전자 돌연변이의 대부분은 APC 유전자의 단백질 생산 이상으로 비정상적인 단백질을 생성하므로 중합효소 연쇄반응과 관내 전사 및 해독 기술(in vitro transcription and translation)을 이용한 절단단백검사(protein truncation test, PTT)도 시행되고 있다. 단백질 검사를 통한 진단은 염기서 열결정법으로 돌연변이가 발견되지 않는 환자의 경우에 있 어서 도움이 될 수 있다.
우리나라에서는 1999년 필자의 연구실에서 가족 용종증 62가계에서 단일쇄형태구조 다형성분석과 절단단백검사를 통하여 38가계(61%)에서 APC 유전자 돌연변이를 발견하여 보고한 바 있다.14
2. 유전 비용종증 대장암
유전 비용종증 대장암(hereditary nonpolyposis colorectal Fig. 2. Development mechanism of fa- milial adenomatous polyposis. Mu- tations in the APC genes that prevent GSK3β-mediated phosphorylation and subsequent β-catenin degradation ulti- mately result in β-catenin pooling within the cellular cytoplasm and nu- cleus. Inside the nucleus, β-catenin associates with members of the T cell factor (TCF) and lymphoid enhancer factor (LEF) family of transcriptional activators. β-catenin and TCF/LEF form a complex that activates trans- cription of target genes.
82 대한소화기학회지: 제45권 제2호, 2005
cancer, HNPCC)은 멘델 우성 유전을 하는 질환으로 대장암 을 비롯하여 각종 암에 걸릴 수 있는 위험성이 증가되는 질 환이다. 전체 대장암의 2-5%를 차지하여 현재까지 알려진 유전 종양 중 가장 빈도가 높다. 1990년 암스테르담에서 유 전 비용종증 대장암 국제협력기구에 의해 국제 공동연구에 서의 통일을 기하기 위하여 최소 진단기준을 정의하였다.15 이러한 진단기준을 통해 많은 예의 유전 비용종증 대장암 환자들이 임상적으로 진단되어 왔지만 이를 위해서는 가계 도 작성에 필요한 정확하고 많은 정보가 필요하고, 유전 비 용종증 대장암과 같이 병발하는 타장기 암에 대한 고려가 없다는 점들이 단점으로 지적되었다. 이러한 단점들을 보완 하고자 1999년 수정된 암스테르담 진단기준(Amsterdam criteria II)이 발표되었는데16 여기에는 대장암 외에 유전 비 용종증 대장암 관련 암으로 자궁내막암, 요관암, 신우암, 소 장암이 포함되었다(Table 3).
1) 유전 비용종증 대장암의 임상 특성
비유전 대장암에 비해 우측 대장에 발생하는 비율이 높 고, 조기에 발병하며, 동시성 및 이시성 대장암의 발생률이 높으면서 대장 이외의 장기에도 암 발생률이 높다. 대장 이 외의 장기에 발생하는 암으로는 자궁내막암과 난소암이 가 장 흔하지만 이 외에도 소장암, 신우암 또는 요관암 등 여러 종류의 암이 한 가계 내에서 동반되어 발생할 수 있다.
유전 비용종증 대장암이 가지는 병리 특징은 분화도가 좋 지 않은 암이나 점액성 암의 발생빈도가 일반 대장암에 비 해 흔하다. 또 이들 환자에서 발견되는 선종은 비교적 조 기에 발생하며 융모 선종이 많고 이형성증이 심하며 일반 적인 선종에 비해 대장암으로 빨리 진행한다. 이러한 조직 소견에도 불구하고 유전 비용종증 대장암 환자의 예후는 일반 대장암 환자보다 좋다. 이는 이 질환이 일반 대장암 보다 조기에 발견된다는 점과 이 질환의 대장암 자체가 일 반 대장암에 비해 덜 침습적이라는 두 가지 이유가 거론되 고 있다.
2) 유전 비용종증 대장암의 원인 유전자
이 질환의 원인이 되는 유전자는 2번 염색체의 단완에 위 치하는데,17 유전 비용종증 대장암 환자의 암 조직에서 분리 한 DNA를 이들 환자의 정상 조직의 DNA와 비교해 보면 단순히 반복되는 염기구조(예를 들어 (CA)n) 길이에 차이가 흔히 발견된다. 이 현상은 현미부수체 불안정(microsatellite instability, MSI)라고 하며 유전 비용종증 대장암의 특징이 다.18 DNA 내의 염기서열 중 단순히 반복되는 염기구조들 은 유전적으로 불안정하기 때문에 유전자의 복제 과정에서 오류가 일어나기 쉬운 부분이다. 이는 유전 비용종증 대장 암에서 암이 발생하는 과정에 수많은 복제 오류가 일어났음 을 의미한다.
이러한 연구 결과를 바탕으로 1993년 2번 염색체의 단완 (2p21-22부위)에서 hMSH2,16 1994년에는 3번 염색체의 단완 (3p21.3)에서 hMLH1,19 2번과 7번 염색체의 hPMS1및 hPMS2 등이 발견되었다.20 이들 유전자들의 공통된 특징은 DNA 복제 시에 발생하는 복제 오류를 교정하는 기능을 가지고 있다는 점이다. 세포분열 시 일어나는 DNA의 정상 복제과 정에서 일정 비율로 오류, 즉 잘못된 염기쌍끼리의 결합이 생겨날 수 있다. 그러나 이러한 잘못된 염기쌍끼리의 결합 부위는 hMSH2와 hMLH1 등의 유전자에 의해 생성되는 단 백질에 의해 결국 올바른 염기쌍으로 치환됨으로써 모든 유 Table 3. Amsterdam Criteria II (revised by International
Collaborative Group on HNPCC, 1999)
At least three relatives with HNPCC-associated cancer (colorectal cancer, cancer of endometrium, small bowel, ureter, or renal pelvis)
1. One should be a first-degree relative of the other two 2. At least two successive generations should be affected 3. At least one should be diagnosed before age 50 years 4. Familial adenomantous polyposis should be excluded
in the colorectal cancer case(s) if any
Tumors should be verified by pathological examination HNPCC, hereditary nonpolyposis colorectal cancer.
Fig. 3. Mismatch-repair pathway. During DNA replication, DNA mismatches may arise, such as from strand slippage or misin- corporation of bases. The mismatch is recognized by MutS homo- logues. MutL homologues are recruited to the complex, and the mismatch is repaired through the action of a number of proteins, including an exonuclease, helicase, DNA polymerase, and ligase.
박재갑 외 1인. 유전 대장암 83
전자들의 기능을 정상적으로 후손 세포들에게 물려주게 된 다. 그러나 hMSH2나 hMLH1 등의 유전자가 돌연변이를 일 으켜 정상 기능을 상실하면 DNA 복제 과정에서 생기는 실 수는 교정되지 못해 후손 세포들의 유전자 기능에 이상이 초래된다. 만일 세포의 분열 및 성장을 조절하는 유전자에 이상이 초래되면 결국 암이 발생하는 것이다21 (Fig. 3). 일반 대장암의 발암 과정은 APC, K-ras, DCC 및 p53 등의 주로 종양억제유전자의 돌연변이가 관여하며 유전자 이상이 순서 대로 일어나는 선종-암종 연속성으로 설명된다. 유전 비용종 증 대장암에서도 역시 선종-암종 연속성을 거치지만 K-ras 돌 연변이가 매우 낮으며, 일반 선종-암종 연속성에서와는 다른 유전자의 변이 과정을 거친다.
복제오류교정유전자의 돌연변이는 90% 이상이 hMSH2와 hMLH1에서 일어나므로 이 두 개의 유전자가 임상적으로 중요하다. 돌연변이는 유전자의 엑손의 전 지역에서 발견되 며 발견되는 돌연변이의 종류는 염기의 결손, 치환, 삽입 등 다양한 형태가 있다. 유전자검사법으로 직접염기서열결정 법은 시간과 비용이 많이 들기 때문에 단일쇄형태구조 다형 성분석 등 보다 간편한 검사법으로 선별 검사를 시행한 후 이상 소견이 보이는 부분에만 적용하는 경향이다.
앞에서 기술한 현미부수체 불안정은 유전이 없는 산발 대 장암 조직에서는 10-15% 정도에서만 발견되지만 유전 비용 종증 대장암에서는 90% 이상에서 발견되어 유전 비용종증 대장암이 의심되는 환자들 중 유전자 진단이 필요한 군을 선별하는 데에 사용할 수 있다. 현미부수체 불안정성검사는 1998년 볼란드 등이 제시한 BAT-25, BAT-26, D5S346, D2S123, D17S250 등 5개의 현미부수체 불안정 표지자(베데 스다 패널) 중 2개 이상의 표지자에서 이상이 있는 군 (MSI-H), 하나의 표지자에서만 이상이 있는 군(MSI-L)과 모 든 표지자에서 이상이 없는 군(MSS)로 나누어 분류하는 것 이22 가장 많이 사용되고 있다.
3) 진단, 검진 및 치료
유전 비용종증 대장암의 진단에는 정확한 가족력을 바탕 으로 한 가계도 분석이 제일 중요하다. 특히 대장암 진단시 연령이 젊거나 이시성 또는 동시성 대장암이 있는 경우, 자 궁내막암 등이 동반된 경우 우선 의심하여야 한다. 진단방 법에는 대장내시경검사가 주로 사용된다. 유전 비용종증 대 장암 가족과 환자를 찾는 목적은 암 발생의 위험도가 높은 가계에 대한 교육과 정기검진을 통한 조기 발견 및 치료, 그 리고 선종에서 암으로 진행되기 전에 선종을 제거하기 위함 이다.
따라서 이 질환에 이환된 가계의 모든 구성원들은 25세부 터 2년마다 대장내시경을 실시하여야 한다. 만일 한 가계 내에 25세 이전에 발생한 대장암 환자가 있는 경우에는 그
환자의 대장암 진단시의 연령보다 5년 어린 나이부터 정기 검진을 시작한다. 35세 이후부터는 대장암 발생 확률이 높 아지므로 매년 대장내시경을 시행하여야 한다.
유전 비용종증 대장암으로 진단되면 수술은 에스결장의 일부와 직장을 제외한 나머지 대장을 절제하는 아전결장절 제술을 시행한다. 또한 여자 환자에 있어서는 결장 절제 후 자궁내막암의 발생 위험도가 높기 때문에 예방 자궁적출술 을 시행하는 것이 좋다.
유전자검사가 환자의 진단, 치료 및 환자의 가족 관리에 대단히 유용한 수단이지만 시간과 비용이 많이 소요되므로 검사를 받아야 할 환자를 선택하는 데 적당한 선별 기준이 있어야 한다. 암스테르담 기준을 만족하는 환자는 의심의 여지없이 유전자검사를 받아야 한다. 암스테르담 기준은 국 제공동연구에서 환자군의 동질성을 유지하는 데는 매우 유 용하지만 수정된 진단기준에서도 그 기준이 너무 엄격하여 실제로 유전자의 이상이 있을 때에도 제외될 수 있다는 단 점이 있다. 이러한 단점을 보완하기 위해 1996년 베데스다 지침이라는 유전자검사의 적응증이 제시되었으며23 이러한
Table 4. Bethesda Guidelines (revised by NIH, 2002) 1. Colorectal cancer diagnosed in a patient who is less
than 50 years of age.
2. Presence of synchronous, metachronous colorectal, or other HNPCC associated tumors, regardless of age.
3. Colorectal cancer with the MSI-H histology diagnosed in a patient who is less than 60 years of age.
4. Colorectal cancer diagnosed in one or more first-degree relatives with an HNPCC-related tumor, with one of the cancers being diagnosed under age 50 years.
5. Colorectal cancer diagnosed in two or more first- or second-degree relatives with HNPCC-related tumors, regardless of age.
HNPCC, hereditary nonpolyposis colorectal cancer; MSI, microsatellite instability.
Table 5. Suspected-HNPCC I Criteria (revised by Park et al.,27 2002, originally proposed by International Collabora- tive Group-HNPCC, 1999)
At least two HNPCC-associated cancers in first-degree relatives (colorectum, endometrium, small intestine, urinary tract), and
1. Multiple colorectal tumors, or
2. At least one HNPCC associated cancer diagnosed be- fore age 50 years, or
3. Development of accompanying cancer in family mem- bers (stomach, biliary, ovary, pancreas)
HNPCC, hereditary nonpolyposis colorectal cancer.
84 The Korean Journal of Gastroenterology: Vol. 45, No. 2, 2005
베데스다 지침에 따라 검사를 시행하였을 때 민감도는 높은 반면, 특이도는 그리 높지 않다.24 2002년에는 기존의 베데 스다 지침을 수정 보완한 수정 베데스다 지침이 발표되었다 (Table 4).25 필자는 암스테르담 진단기준에는 해당되지 않지 만 유전자검사가 필요한 경우로 유전의심 비용종증 대장암 (suspected-HNPCC) 환자의 기준을 정하고(Table 5) 국제공동 연구를 시행하였다.26,27 연구 결과 수정된 암스테르담 기준 을 만족하는 경우 50%에서 돌연변이가 발견된 데 비해 수 정된 유전의심 비용종증 기준에서도 26%에서 돌연변이가 발견되어27 유전의심 대장암의 경우에도 유전자검사가 필요 함을 확인하였다. 최근 한국유전종양등록소에서는 지금까 지 등록된 유전 비용종증 대장암 가계 및 유전의심 비용종 증 대장암 가계에 대한 유전자검사 결과를 이전의 보고들
과28-31 같이 보고한 바 있는데 이에 따르면 53가족의 유전
비용종증 대장암 가계 중 22가족(41.5%)에서 복제오류교정 유전자의 배선돌연변이(hMLH1 91%, hMSH2 4.5%, hMSH6 4.5%)가 발견되었으며 111가족의 유전의심 비용종증 대장 암 가계 중 22가족(19.8%)에서 복제오류교정유전자의 배선 돌연변이가 발견되었다.32 돌연변이가 발견된 44가족 중 11 가족은 동일한 돌연변이(c.1757_1758insC)를 가지고 있었으 며, 이들 가족에 대한 haplotype 분석 결과 이 돌연변이는 동 일한 조상에서 기원한 개척자 돌연변이(founder mutation) 임을 확인할 수 있었다.32
3. 포이츠-예거스 증후군
포이츠-예거스 증후군은 멘델 우성 유전을 하는 질환으로 위장관에 과오종을 형성하고 피부점막부에 색소 침착이 특 징적인 질환이다. 빈도는 가족 용종증이나 유전 비용종증 대장암에 비해 훨씬 드물다. 과오종 자체가 악성 종양으로 발전하는 경우는 드물지만 이 질환을 가진 가족에서 대장암
이 다발하는 증례들이 보고되어 있다. 대장암 외에도 유방 암, 자궁경부암, 고환암 및 췌장암 등이 호발하며 양성 종양 으로는 위장관의 과오종 이외에 비강, 기관지, 요로계 등에 도 과오종을 유발한다. 염색체 19p13.3에 위치하는 STK11 유전자의 배선돌연변이가 포이츠-예거스 증후군의 원인 유 전자로 발견되었다.33 우리나라에서는 2000년 필자의 연구 실에서 10명의 포이츠-예거스 증후군 환자들 중 5예에서 STK11 유전자의 돌연변이를 발견하여 보고한 바 있다.34 이러한 포이츠-에거스 증후군 환자들에 대한 치료는 예방 장 절제는 권유되지 않으며 암 발생 시에는 통상적인 대장 절제를 시행하고 과오종에 대해서는 증상과 크기에 따라 수 술 절제 또는 내시경 절제를 시행한다.
4. 연소기 용종증
연소기 용종증은 비교적 최근에 알려진 드문 질환으로 멘 델 우성 유전을 한다. 이 질환은 포이츠-예거스 증후군과 마 찬가지로 위장관에 과오종을 가지며 악성으로 전환을 하지 는 않는 것으로 알려져 왔으나 최근에는 일부 용종은 선종 양상을 갖고 악성으로 전환된다. 수술 치료법은 가족 용종 증과 마찬가지로 전대장절제술 및 회장낭-항문문합술을 시 행하는 것이 추천된다.
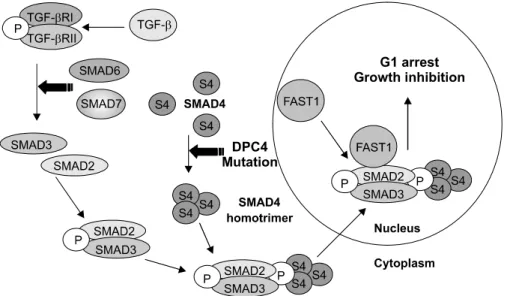
이 질환의 원인 유전자는 1988년 호웨 등에 의해 SMAD4 (DPC4)가 원인 유전자임이 밝혀졌다.35 SMAD4는 18q에 위 치하며 종양억제유전자로 알려진 DCC와 매우 근접한 위치 에 존재한다. SMAD4는 TGF-β의 세포 내 신호전달체계에 관여하는 단백질을 부호화하는 유전자로 552개의 아미노산 으로 이루어진 단백을 생산한다. 체내에서 SMAD4 유전자의 돌연변이가 발생하면 TGF-β 신호전달체계에서 SMAD4 단 백질의 동종 삼합체(homotrimer) 형성이 이루어지지 않아 핵 내 세포성장억제신호가 저해된다(Fig. 4). SMAD4 유전자의
Fig 4. TGF-β signaling pathway and SMAD4 (DPC4). TGF-beta regulates growth and proliferation of cells. The TGF-beta receptor includes type 1 and type 2 subunits that signal th- rough the SMAD family of trans- criptional regulators. Defects in TGF- beta signaling have been associated with cancer in humans. Binding of TGF induces phosphorylation and ac- tivation of the TGF-beta R1 by the TGF-beta R2. The activated TGF- beta R1 phosphorylates SMAD2 and SMAD3, which bind to the SMAD4 mediator to move into the nucleus and form complexes that regulate transcription.
Park JG, et al. Hereditary Colorectal Cancer 85
체성돌연변이는 췌장암에서 50%, 대장암에서 15% 정도로 나타나, 연소기 용종증에서 흔히 발견되는 췌장암 및 대장 암의 생성에 중요한 역할을 할 것으로 여겨진다.
SMAD4 유전자 돌연변이가 없는 4명의 환자들에서 BMPR1A (bone morphogenetic protein receptor 1A) 유전자의 돌연변이 가 관찰되어 연소기 용종증의 원인 유전자로 SMAD4 유전 자 외에 BMPR1A 유전자가 관여한다.36 필자의 연구실에서 는 우리나라 연소기 용종증 환자들의 유전자검사를 시행하 여, 연소기 용종증 환자 4명 중 3예에서는 DPC4 유전자의 배선돌연변이, 1예는 BMPR1A 유전자의 배선돌연변이를 발 견하였다.37,38
5. MYH 용종증
2002년 알-타산 등은 다발 대장선종과 암종이 형제, 자매 사이에 집중된 영국인 가계(Family N)를 연구하였는데, 이 가계의 구성원들의 APC 유전자검사에서 배선돌연변이는 없으나 다만 구아닌-사이토신 염기쌍이 티민-아데닌 염기쌍 으로 변이(transversion)된 체성돌연변이만이 존재하는 것을 발견하였고, 이 결과를 토대로 염기절제교정(base excision repair)에 관여하는 복제교정유전자인 MYH 유전자의 배선 돌연변이를 최초로 보고하였다.5 염기절제교정유전자는 체 내에서 산화성 DNA 손상 시 발생하는 8-옥소 구아닌이 사 이토신 대신 아데닌과 결합하는 결합 오류를 방지 또는 교 정하는 작용을 하는 유전자로 생체 내에는 OGG1, MYH, MTH1의 세 가지 유전자가 있다. 이 중 MYH 유전자는 아데
닌과 잘못 결합되어 있는 옥소 구아닌을 사이토신과 결합하 도록 교정하는 역할을 한다(Fig. 5).
이러한 MYH 용종증은 가족 용종증과 같이 다발 용종이 발생되고 대장암의 발생 위험도가 높으며 임상상으로는 가 족 용종증의 경한 유형(attenuated FAP)과 유사하나 대부분 의 유전 종양들과는 달리 두 개의 대립유전자 모두에서 MYH 돌연변이가 발견되는 멘델 열성 유전을 한다. 발생빈 도는 아직 명확하지 않으나 APC 유전자의 배선돌연변이가 없는 가계들 중 선종이 100개 이상인 경우 7%, 선종이 100 개 이하 15개 이상인 경우 약 30%에서 MYH 유전자의 배선 돌연변이가 보고되었다.39 이들에서의 선종과 암종의 분자 생물학 특성은 K-ras 유전자 돌연변이가 빈번하고 p53 돌연 변이는 드물게 발견되나 현미부수체 불안정은 없다.40
참고문헌
1. Shin HR, Jung KW, Won YJ, Park JG. 2002 annual report of the Korea Central Cancer Registry; based on registered data from 139 hospitals. Cancer Res Treat 2004;36: 103-114.
2. Trimbath JD, Giardiello FM. Genetic testing and counselling for hereditary colorectal cancer. Aliment Pharmacol Ther 2002;16:1843-1857.
3. Park JG. Human and Hereditary Disease. Seoul; Dong-a Pub- lishing Co., 1995
4. Cannon-Albright LA, Skolnick MA, Bishop T, Lee RG, Burt RW. Common inheritance of susceptibility to colonic adeno- Fig. 5. Base excision repair (BER) pathway. Human cellular genomes suffer damage from reactive oxygen species formed during normal metabolism. The MTH remove oxidized nucleotide precursors so that they cannot be incorporated into DNA during replication. The 8-oxoG glycosylases (OGG) and the MYH glycosylases along with MTH protect cells from the mutagenic effects of 8-oxoG, the most stable and deleterious product known caused by oxidative damage to DNA. The OGG1 glycosylase initiates base excision repair (BER) of 8-oxoguanine (8-oxoG) from 8-oxoG:C pairs. The MYH glycosylase removes mismatched adenines incorporated opposite 8-oxoG during replication. Subsequent BER generates 8-oxoG:C pairs, a substrate for excision by OGG1.
86 대한소화기학회지: 제45권 제2호, 2005
matous polyps and associated colorectal cancers. N Engl J Med 1988;319:533-537.
5. Al-Tassan, Chmiel NH, Maynard J, et al. Inherited variants of MYH associated with somatic G:C→T:A mutations in colorectal tumors. Nat Genet 2002;30:227-232.
6. Rustin RB, Jagelman DG, McGannon E, Fazio VW, Lavery IC, Weakley FL. Spontaneous mutation in familial adeno- matous polyposis. Dis Colon Rectum 1990;33:52-55.
7. Bussey HJ. Familial polyposis coli. Family studies, histopa- thology, differential diagnosis and results of treatment. Balti- more; Johns Hopkins University Press, 1975.
8. Park JG, Park KJ, Won CK, et al. Polyposis coli syndrome in Koreans (1990) - Korean Polyposis Registry. J Korean Soc Coloproctol 1991;7:1-13.
9. Tonelli F, Valanzano R, Messerini L, Ficari F. Long-term treatment with sulindac in familial adenomatous polyposis: is there an actual efficacy in prevention of rectal cancer? J Surg Oncol 2000;74:15-20.
10. Steinbach G, Lynch PM, Phillips RK, et al. The effect of celecoxib, a cyclooxygenase-2 inhibitor, in familial adenoma- tous polyposis. N Engl J Med 2000;342:1946-1952.
11. Bodmer WF, Bailey CJ, Bodmer J, et al. Localization of the gene for familial adenomatous polyposis on chromosome 5.
Nature 1987;328:614-616.
12. Groden J, Thliveris A, Samowitz W, et al. Identification and characterization of the familial adenomatous polyposis coli gene. Cell 1991;66:589-600.
13. Park JG, Han HJ, Kang MS, Nakamura Y. Presymptomatic diagnosis of familial adenomatous polyposis coli. Dis Colon Rectum 1994;37:700-707.
14. Won YJ, Park KJ, Kwon HJ, et al. Germline mutations of the APC gene in Korean familial adenomatous polyposis patients.
J Hum Genet 1999;44:103-108.
15. Vasen HF, Mecklin JP, Khan PM, Lynch HT. The Inter- national Collaborative Group on Hereditary Nonpolyposis Colorectal Cancer (ICG-HNPCC). Dis Colon Rectum 1991;
34:424-425.
16. Vasen HFA, Watson P, Mecklin J-P, Lynch HT. New clini- cal criteria for hereditary non polyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative Group on HNPCC. Gastroenterology 1999;116:
1453-1456.
17. Fishel R, Lescoe MK, Rao MR, et al. The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell 1993;75:1027-1038.
18. Peltomaki P, Lothe RA, Aaltonen LA, et al. Microsatellite instability is associated with tumors that characterize the
hereditary nonpolyposis colorectal carcinoma syndrome. Can- cer Res 1993;53:5853-5855.
19. Bronner CE, Baker SM, Morrison PT, et al. Mutation in the DNA repair gene homologue hMLH1 is associated wtih hereditary non-polyposis colon cancer. Nature 1994;368:258- 261.
20. Nicolaides NC, Papadopoulus N, Liu B, et al. Mutations of two PMS homologues in hereditary nonpolyposis colon can- cer. Nature 1994;371:75- 80.
21. Han HJ, Maruyama M, Baba S, Park J-G, Nakamura Y.
Genomic structure of human mismatch repair gene hMLH1, and its mutation analysis in patients with hereditary non- polyposis colorectal cancer (HNPCC). Hum Mol Genet 1995;
4:237-242,
22. Boland CR, Thibodeau SN, Hamilton SR, et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res 1998;58:5248- 5257.
23. Rodriguez-Bigas MA, Boland CR, Hamilton SR, et al. A National Cancer Institute Workshop on Hereditary Nonpoly- posis Colorectal Cancer Syndrome: meeting highlights and Bethesda guidelines. J Natl Cancer Inst 1997;89:1758-1762.
24. Terdiman JP, Gum JR Jr, Conrad PG, et al. Efficient detection of hereditary nonpolyposis colorectal cancer gene carriers by screening for tumor microsatellite instability be- fore germline genetic testing. Gastroenterology 2001;120:21- 30.
25. Umar A, Boland CR, Terdiman JP, et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst 2004;96:261-268.
26. Park JG, Vasen HF, Park KJ, et al. Suspected hereditary nonpolyposis colorectal cancer: International Collaborative Group on Hereditary Non-Polyposis Colorectal Cancer (ICG- HNPCC) criteria and results of genetic diagnosis. Dis Colon Rectum 1999;42:710-715.
27. Park JG, Vasen HF, Park YJ, et al. Suspected HNPCC and Amsterdam criteria II:evaluation of mutation detection rate, and international collaborative study. Int J Colorectal Dis 2002:17:109-114.
28. Han HJ, Yuan Y, Ku JL, et al, Germline mutations of hMLH1 and hMSH2 genes in Korean hereditary nonpolyposis colorectal cancer. J Natl Cancer Inst 1996;88:1317-1319.
29. Yuan Y, Han HJ, Zheng S, Park JG. Germline mutations of hMLH1 and hMSH2 genes in patients with suspected her-
박재갑 외 1인. 유전 대장암 87
editary nonpolyposis colorectal cancer and sporadic early- onset colorectal cancer. Dis Colon Rectum 1998;41:434-440.
30. Shin KH, Ku JL, Park JG. Germline mutations in a poly- cytosine repeat of the hMSH6 gene in Korean hereditary nonpolyposis colorectal cancer. J Hum Genet 1999;44:18-21.
31. Shin KH, Shin JH, Kim JH, Park JG. Mutational analysis of promoters of mismatch repair genes hMSH2 and hMLH1 in hereditary nonpolyposis colorectal cancer and early onset colorectal cancer patients: identification of three novel germ- line mutations in promoter of the hMSH2 gene. Cancer Res 2002;62:38-42.
32. Shin YK, Heo SC, Shin JH, et al. Germline mutations in MLH1, MSH2 and MSH6 in Korean hereditary non-polyposis colorectal cancer families. Human Mutat 2004;24:351.
33. Jenne DE, Reimann H, Nezu J, et al. Peutz-Jeghers syndrome is caused by mutations in a novel serine threonine kinase.
Nat Genet 1998;18:38-43.
34. Yoon KA, Ku JL, Choi HS, et al. Germline mutations of the STK11 gene in Korean Peutz-Jeghers syndrome patients. Br J Cancer 2000;82:1403-1406.
35. Howe JR, Roth S, Ringold JC, et al. Mutations in the SMAD4/DPC4 gene in juvenile polyposis. Science 1998;280:
1086-1088.
36. Howe JR, Bair JL, Sayed MG, et al, Germline mutations of the gene encoding bone morphogenetic protein receptor 1A in juvenile polyposis. Nat Genet 2001;28:184-187.
37. Kim IJ, Ku JL, Yoon KA, et al. Germline mutation of the dpc4 gene in Korean juvenile polyposis. Int J Cancer 2000;
86:529-532.
38. Kim IJ, Park JH, Kang HC, et al. Identification of a novel BMPR1A germline mutation in a Korean juvenile polyposis patient without SMAD 4 mutation. Clin Genet 2003;63:126- 130.
39. Sieber OM, Lipton L, Crabtree M, et al. Multiple colorectal adenomas, classic adenomatous polyposis, and germ-line mu- tations in MYH. N Engl J Med 2003;348:791-799.
40. Lipton L, Halford SE, Johnson V, et al. Carcinogenesis in MYH-associated polyposis follows a distinct genetic pathway.
Cancer Res 2003;63:7595-7599.