514 REVIEW
Korean Circ J 2008;38:514-523
Print ISSN 1738-5520 / On-line ISSN 1738-5555 Copyright ⓒ 2008 The Korean Society of Cardiology
Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia
Yongkeun Cho, MD
Department of Internal Medicine, School of Medicine, Kyungpook National University, Daegu, Korea ABSTRACT
Arrhythmogenic right ventricular cardiomyopathy/dysplasia (ARVC/D) is characterized by progressive, fibrofatty replacement of the myocardium, ventricular arrhythmia, sudden death, and progressive heart failure. ARVC/D may be an important cause of syncope, ventricular arrhythmias, electrocardiogram (ECG) abnormalities and/or non-ischemic wall motion abnormalities. Some patients, however, do not have a typical clinical presentation. Thus, a high clinical suspicion and extensive studies may be needed to establish the diagnosis of ARVC/D. Recent progress in diagnostic modalities and a better understanding of the clinical manifestations of ARVC/D may lead to optimal management of affected patients. (Korean Circ J 2008;38:514-523)
KEY WORDS: Cardiomyopathies; Arrhythmia; Right ventricle.
Introduction
Arrhythmogenic right ventricular cardiomyopathy/
dysplasia (ARVC/D) is a primary disease of heart muscle characterized by progressive, fibrofatty replacement of the right ventricle (RV) and life-threatening ventricular arrhythmias with left bundle branch block (LBBB) mor- phology.1-8) Twenty-six years have elapsed from the time that the clinical profile of ARVC/D was first described.1) ARVC/D in animals, such as cats and dogs, have also been reported.9)10)
The estimated prevalence of ARVC/D in the general population ranges from 1 in 2,000 to 1 in 5,000.11) This condition was initially believed to be a developmental defect of the RV myocardium, leading to the original designation of ‘dysplasia’. This concept has evolved over the last 25 years into the current perspective of a gene- tically-determined ‘cardiomyopathy.’
Etiology
Several reports have suggested a familial pattern for ARVC/D;1)12)13) however, the precise etiology of ARVC/
D remains to be established. ARVC/D is typically in- herited as an autosomal dominant trait with incomplete penetrance and variable expression,12)13) implicating en- vironmental factors and other genetic modifiers in the
etiology of this disease. An autosomal recessive form as- sociated with palmoplantar keratosis is called Naxos disease. The locus for Naxos disease has been located on chromosome 17q21.14) A mutated gene produces a pro- tein called plakoglobin is involved in the genesis of the disease.15) A recessive mutation in the desmoplakin gene has also been described as the etiology of Naxos disease.16) Disease loci for the autosomal dominant form of AR- VC/D have been mapped to chromosomes 14q23-24, 1q42-q43, 14q12-q22, 2q32, 3q23, 10p12-p14, 10q22, and 6p24.17-22) Apoptosis has also been proposed as the underlying mechanism in the pathogenesis of this dis- ease.23)24)
Incidence and Prevalence
The incidence is unknown since this pathologic process can be completely asymptomatic in the young, with sud- den death as the initial manifestation.5)7)25) However, a prevalence of approximately 1 in 5,000 people has been estimated.11) ARVC/D has been found in up to 20% of sudden deaths in young people and it is considered the most common cause of exercise-related sudden death among young Italian athletes.25) The incidence of a fa- milial pattern varies from 15-50% with the study po- pulation and method.12)13)26)27)
Natural History
Classically, the natural history of ARVC/D is con- sidered to include four distinct phases. In the first phase, the concealed phase, patients are often asymptomatic,
Correspondence: Yongkeun Cho, MD, Department of Internal Medicine, School of Medicine, Kyungpook National University, 50 Samdeock 2-ga, Jung-gu, Daegu 700-721, Korea
Tel: 82-53-420-5528, Fax: 82-53-426-2046 E-mail: [email protected]
Yongkeun Cho·515
but may nevertheless be at risk for sudden death, es- pecially during exertion. Structural changes are subtle and may be confined to one region of the so-called
“triangle of dysplasia:” (the inflow, outflow, and apical portions of the RV). Symptomatic ventricular arrhythmia is seen in the second phase, the overt electrical disorder phase, accompanied by more obvious morphologic and functional abnormalities of the RV. The third phase is the RV failure phase is characterized by diffuse RV disease, which results in right-sided heart failure with relatively preserved left ventricular (LV) function. Notable LV involvement with biventricular pump failure occurs in the fourth biventricular pump failure stage, leading to a phenotype that may resemble dilated cardiomyopa- thy.28) A multicenter study of patients with a pathologic diagnosis of ARVC/D at transplantation or autopsy found evidence of fibrofatty replacement of the LV in more than two-thirds of the hearts examined.5) LV in- volvement correlated with age, arrhythmic events, and clinical heart failure.
Another recent European study described the annual mortality rate of 2.3%.29) Progressive heart failure was the cause of death in more than one-half of patients, whereas sudden death occurred in less than one-third of patients. In this series, the high prevalence of death due to heart failure may be explained by a change in the cause of death from sudden death to non-sudden death due to aggressive therapeutic management, including ablation, implantable cardioverter-defibrillator (ICD), and surgical treatment. In another study of family mem- bers, the yearly mortality rate was very low (0.08 patient per year).
These data illustrate the wide spectrum of the natural history of the disease, and the presence of subgroups of patients with variable degrees of risk.
Pathology
Dilatation of the RV, aneurysmal dilatations of the infundibular, apical and subtricuspid areas, and repla- cement of normal myocardium with fibrofatty tissue and thinning of the RV wall are commonly observed in cardiac tissues obtained from patients with ARVC/
D.1)2)5)12)30) The interventricular septum and the LV are spared until late stages of the disease. As the pathologic process advances from the epicardium to the endocar- dium, an endocardial biopsy obtained from a suspected case may be normal. Two types of ARVC/D, fatty and fibrofatty types, were proposed 30 years ago.2) However, pure fatty infiltration of the RV, per se, is a different process that may not be considered synonymous with ARVC/D.30) The diagnosis of ARVC/D should be made in the presence of fibrosis, which is more arrhythmo- genic than pure fatty infiltration.30)31) The other histo- logic finding which exists in patients with ARVC/D is
lymphocytic infiltration of the myocardium.5) Younger patients dying with fibrofatty ARVC/D may have a more lethal or aggressive form of the disease, characterized by myocardial inflammation. Another controversy which persists regarding ARVC/D is the clinical significance of conduction abnormalities, as reported in a French study.32)
Symptoms
The clinical presentation varies from silent forms with an exercise-related episode of syncope or sudden death as the initial manifestation, to biventricular failure that requires cardiac transplantation.1)2)5-8) RV failure is usually observed in older patients. Common symptoms include palpitations, fatigue, dizziness, atypical chest pain, and syncope.8)33) The mechanism of sudden death in ARVC/
D is known to be an acceleration of ventricular tachy- cardia (VT) with ultimate degeneration into ventricular fibrillation (VF).29) ARVC/D should be suspected in all young patients presenting with syncope, VT, or cardiac arrest.7)25)31) Patients with ARVC/D most commonly come to clinical attention because of ventricular arrhy- thmias. The ventricular arrhythmias that originate in the diseased RV may be asymptomatic and detected by routine ECG, or they may cause symptoms, such as palpitations, syncope, or sudden cardiac death. Exercise has been identified as a common precipitant of arrhyth- mias that occur in ARVC/D.5)11)25)34)
Physical Examination
About one-half of patients have normal findings on physical examination. With severe RV dilation, tricuspid regurgitation murmur and giant a waves may be ob- served.33)
Diagnosis
In 1994, the Task Force of the Working Group on Cardiomyopathies proposed diagnostic criteria for AR- VC/D (Table 1).35) This task force was established be- cause the diagnosis of ARVC/D may be difficult due to several problems, such as non-specific ECG abnormal- ities, the diverse potential etiologies of ventricular arrhy- thmias with a LBBB morphology, the technical difficul- ties in assessing RV structure and function, and the interpretation of the endomyocardial biopsy findings.
The diagnosis of ARVC/D is fulfilled when two major criteria or one major criterion plus two minor criteria or four minor criteria exist from different groups. The criteria are highly specific, but lack sensitivity. Diagnosis at an early stage remains a clinical challenge. The eerial evaluation of patients with suspected ARVC/D is re- commended as clinical features may develop during the
516·Arrhythmogenic RV Cardiomyopathy/Dysplasia
follow-up period.13)27)33)
It has been recommended that patients suspected to have ARVC/D undergo a thorough initial evaluation with non-invasive testing. Standard non-invasive testing for ARVC/D includes a 12-lead ECG, echocardiography, signal-averaged ECG, Holter monitoring, and cardiac magnetic resonance imaging. If non-invasive testing re- veals findings consistent with ARVC/D, then invasive testing, including RV angiography and a RV endomyo- cardial biopsy, are recommended to confirm the diag- nosis.36)
A definite diagnosis of ARVC/D is based on histologic demonstration of transmural fibrofatty replacement of the RV myocardium obtained at autopsy or surgery.
Myocardial biopsy lacks sufficient sensitivity to establish the diagnosis because the biopsy is performed mostly in the interventricular septum, whereas the typical patho- logic changes of ARVC/D are more pronounced in the RV free wall.31) It is important not to rely on any single criteria to arrive at a diagnosis of ARVC/D, particularly imaging studies. All reasonable efforts should be made to firmly establish the diagnosis.
Routine ECG
ECG abnormalities are detected in most patients with ARVC/D (Table 2).8)29)37-39) T-wave inversion beyond V1
in a young- or middle-aged patient with no apparent
Table 1. Task Force of the Working Group on Cardiomyopathies diagnostic criteria for arrhythmogenic right ventricular cardiomyopa- thy/dysplasia35)
Major criteria Minor criteria
I Global and/or regional dysfunction and structural alterations
Severe dilation and reduction of right ventricular ejection fraction with no (or only mild) left ventricular impairment
Mild global right ventricular dilation and/or ejection fraction reduction with normal left ventricle
Localized right ventricular aneurysms (akinetic or dyskinetic areas with diastolic bulging)
Mild segmental dilation of the right ventricle Severe segmental dilation of the right ventricle Regional right ventricular hypokinesia II Tissue characterization of walls Fibrofatty replacement of myocardium on
endomyocardial biopsy
III Repolarization abnormalities Inverted T waves on right precordial leads (V2 and V3) (age >12 years; in absence of right bundle branch block)
IV Depolarization/Conduction abnormalities
Epsilon waves or localized prolongation (>110 ms) of the QRS complex in right precordial leads (V1-V3)
Late potentials (signal-averaged ECG)
V Arrhythmias Left bundle branch block-type ventricular
tachycardia (sustained and nonsustained) (ECG, Holter, exercise testing)
Frequent ventricular extrasystoles (>1,000/24 hours) (Holter)
VI Family history Familial disease confirmed at necropsy or surgery Familial history of premature sudden death (<35 years) due to suspected right ventricular dysplasia Familial history (clinical diagnosis based on present
criteria)
To fulfill the appropriate criteria for arrhythmogenic right ventricular dysplasia/cardiomyopathy, patients must meet either two major criteria, one major plus two minor criteria, or four minor criteria
Table 2. Common ECG features in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia39)
Electrocardiographic pattern Reported prevalence in probands (Varies by Cohort)
Normal Up to 40% of patients with symptomatic ventricular tachycardia and
right ventricular dysplasia in first year of follow-up Depolarization abnormalities
Complete right bundle branch block 6% to 15%
Incomplete right bundle branch block 14% to 18%
Prolongation of QRS complex to >110 ms in V1-V3 in absence of right bundle branch block*
Up to 98% of patients fulfilling task force criteria for right ventricular dysplasia
Right precordial R-wave reduction 5%
Epsilon waves in right precordial leads* 23% using standard recording technique; 75% when highly amplified and modified recording techniques are employed in adjunct Late potentials on signal-averaged ECG 50% to 80%
Repolarization abnormalities
Inverted T waves in V1-V3 in absence of right bundle branch block*
54%
*abnormalities cited in the task force criteria
Yongkeun Cho·517
heart disease, but with ventricular arrhythmias of LBBB morphology, should raise the suspicion of ARVC/D.
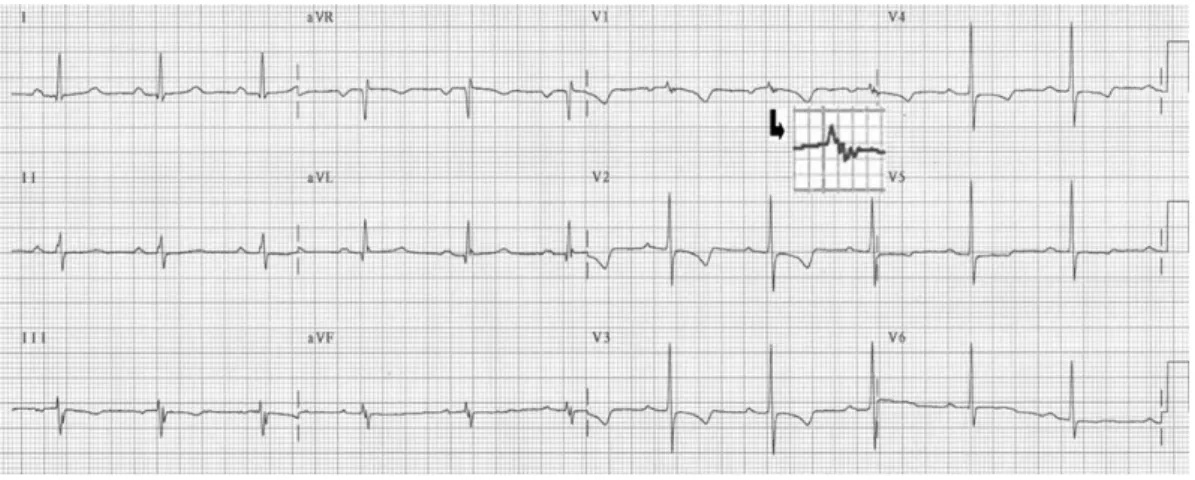
Another typical ECG feature of ARVC/D is the so-call- ed epsilon waves, which are post-excitation electrical potentials of small amplitude that occur at the end of the QRS complex and at the beginning of the ST segment (Fig. 1). Epsilon waves, which are seen in one-third of patients, are considered a major diagnostic criterion for ARVC/D. Slowed electrical conduction in the RV as a result of ARVC/D may also cause localized widening of QRS complex (≥110 ms) in the right precordial leads, which is seen in two-thirds of the patients. A prolonged S-wave upstroke in V1 through V3 ≥55 ms is the most prevalent ECG feature in most patients and is correlat- ed with disease severity and induction of VT.11)29)38)39)
Chest X-Ray
The chest X-ray is usually normal, unless RV enlarge- ment and failure are present.
Signal-Averaged ECG
Late potentials on SAECG recordings are considered a minor criterion for the diagnosis of ARVC/D.35) Us- ually, when ≥2 of the following criteria are present, late potentials are considered to be present: (1) the duration of the signal-averaged, high-frequency filtered QRS com- plex ≥114 ms, (2) the duration of the low-amplitude signal of <40 μV in the terminal portion of the filtered QRS complex ≥38 ms, and (3) the root mean square voltage of the terminal 40 ms of the filtered QRS com- plex <20 μV. An abnormal signal-averaged ECG was present in about two-thirds of the patients,8)29) and some of the family members.26)
Exercise ECG
A symptom-limited exercise stress test is usually per-
formed. Frequent premature ventricular contractions (PVCs), non-sustained VT, and/or sustained VT with exercise were considered abnormal.35)36)
Holter Monitoring
Holter monitoring is considered abnormal when
>1,000 PVCs are present in 24 hours.35) The presence of ventricular arrhythmias represents a particularly frequent and characteristic form of presentation.8)33) VF and sud- den death can be observed during exercise or physical activity.25)40) However, episodes of sudden death during sleep also occur in patients with normal physical exa- minations.7)33) Supraventricular arrhythmias, including atrial premature contractions, flutter, tachycardia, and fibrillation have been reported in one-third of patients with ARVC/D and ventricular arrhythmias.41)
Imaging of the RV
RV size and function can be evaluated using a variety of imaging modalities, including 2-D echocardiography, contrast angiography, cardiac magnetic resonance imag- ing (MRI), and computed tomography (CT). The pre- sence of severe dilatation and/or functional loss of the RV are considered major criterion, and a less severe abnormality in RV size and/or function is considered to be a minor criterion for ARVC/D. However, the de- finitions of severe and non-severe are unclear and the diagnostic accuracy of each test is uncertain.
RV Contrast Angiography
RV angiography has been regarded as the gold stand- ard for the diagnosis of ARVC/D and has been shown to be highly specific.11) Dyskinetic or akinetic zones in the infundibular, apical, or subtricuspid regions are highly specific findings for ARVC/D; increased end- diastolic volume with abnormal wall motion, persistence
Fig. 1. Routine ECG in a patient with arrhythmognic right ventricular cardiomyopathy/dysplasia showing diffuse precordial T-wave inversion in the right precordial leads and an epsilon wave (arrow) in lead V1.
518·Arrhythmogenic RV Cardiomyopathy/Dysplasia
of contrast, multiple outpouchings of the inferior RV wall or aneurysms, tricuspid or mitral valve prolapse, isolated diastolic protrusions, and systolic dyskinesia of the outflow tract are also found. Although the RV volume is nearly always increased, it is often difficult to assess RV enlargement because of the complex geometric shape of the RV.
The invasive nature of the angiographic technique, radiation exposure, and considerable interobserver va- riability regarding the visual assessment of RV motion abnormalities preclude the use of RV angiography as a primary diagnostic technique for ARVC/D.31)
Echocardiography
Echocardiography is the most widely used and sensitive technique for assessing cardiac performance in patients with ARVC/D and their family members.8)31)42) The most conspicuous findings on echocardiography are RV dila- tion, enlargement of the right atrium, isolated dilation of the RV outflow tract, increased reflectivity of the mo- derator band, localized aneurysms, and decreased frac- tional area change (Fig. 2).42) RV outflow dilatation is the most common abnormality associated with the task force diagnostic criteria for ARVC/D.42)
Cardiovascular Magnetic Resonance Imaging and Computed Tomography
One of the most important advances in the diagnosis of ARVC/D has been the introduction of MRI. Non- invasive detection of RV myocardial fibrofatty changes in ARVC/D was made possible by MRI (Fig. 3). MRI findings have an excellent correlation with histopathology and predicted inducible VT, suggesting a possible role in the evaluation and diagnosis of patients with sus- pected ARVC/D.43)
However, limitations of MRI, including interobserver variability, lack of experience at most centers, insuffi- cient resolution for detection of wall thinning, overin- terpretation of regional wall motion abnormalities, and difficulties in differentiating normal epicardial fat from true myocardial adipose replacement are known. Thus, the routine use of MRI should not be performed unless an experienced MRI center is available.11)
MRI even leads to misdiagnosis of ARVC/D and un- necessary ICD implantation. Diagnosis of ARVC/D cannot rely solely upon quantitative features on MRI.36) Cardiac MR is a valuable component of the diagnostic workup for ARVC/D when performed with a dedicated protocol by experienced specialists.44)
CT demonstrates an increase in epicardial adipose tissue delineated by densitometric analysis of the image.
This test also shows other morphologic abnormalities described above, such as localized or diffuse compromise, dilatation of the RV, thinning of the wall, hypokinesis, and prominent trabeculations with low attenuation.
Multi-detector CT may be an alternative to MRI for pa- tients in whom MRI is a contraindication because of the presence of a pacemaker or ICD.45)
Endomyocardial Biopsy
Endomyocardial biopsy is recommended for all pa-
Fig. 2. Two-D echocardiography showing localized (A) and dif- fuse (B) dilatation of the right ventricle in patients with arrhyth- mogenic right ventricular cardiomyopathy/dysplasia.
A
B
Fig. 3. Magnetic resonance imaging of a patient with arrhyth- mogenic right ventricular cardiomyopathy/dysplasia. The right ventricle was dilated and fatty infiltration of the right ventricular free wall, apex, and left ventricular apex was observed.
Yongkeun Cho·519
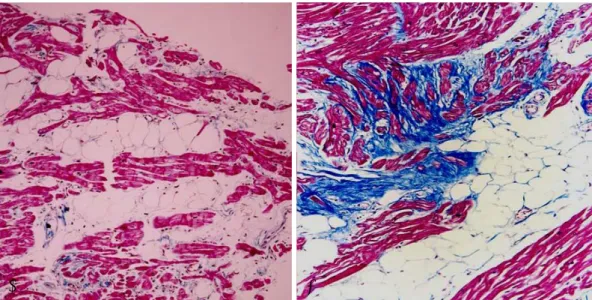
tients suspected of having ARVC/D. It is important to recognize that ARVC/D can be a patchy disease and that clear evidence for the diagnosis will be obtained in ap- proximately one-third of affected individuals. However, when the biopsy shows fibrosis and fatty infiltration, along with surviving strands of myocytes, the diagnosis of ARVC/D is clearly established (Fig. 4). An endomyo- cardial biopsy is also useful in excluding other condi- tions, such as sarcoidosis and myocarditis, which can be confused with ARVC/D.11)46)
Gene Study
A gene study is not feasible in the majority of cases because of the large genetic heterogeneity and limitation of molecular screening to only a handful of genes that account for minor variants of the disease (Table 3). The- refore, genetic analysis is not currently available for the
clinical diagnosis of ARVC/D and is restricted to re- search laboratories.
Genetic analysis is useful in families with ARVC/D because whenever a pathogenetic mutation is identified, it becomes possible to establish a presymptomatic diag- nosis of the disease among family members, to provide them with genetic counseling to monitor the develop- ment of the disease, and to assess the risk of transmitting the disease to offspring. On the basis of current knowl- edge, genetic analysis does not contribute to risk stra- tification of ARVC/D.47)
Electrophysiologic Study
The prognostic role of an electrophysiologic study (EPS) in patients with ARVC/D is controversial. In an Italian study, the limited value of EPS in identifying pa- tients at risk has been demonstrated.40) However, in an
Table 3. Summary of genes associated with arrhythmogenic right ventricular cardiomyopathy/dysplasia47)
Gene Coding protein Chromosome location Mode of transmission
ARVD1 TGFB3 TGFB3 14q24 AD
ARVD2 RYR2 Ryanodine receptor 1q42-q43 AD
ARVD3 ? ? 14q12-q22 AD
ARVD4 ? ? 2q32.1-q32.3 AD
ARVD5 TMEM43 Transmembrane protein 3p23 AD
ARVD6 ? ? 10p12-p14 AD
ARVD7 ? ? 10q22.3 AD
ARVD8 DSP Desmoplakin 6p24 AD/AR
ARVD9 PKP2 Plakophilin 12p11 AD/AR
ARVD10 DSG2 Desmoglein 18q12.1-q12.2 AD
ARVD11 DSC2 Desmocollin 18q12.1 AD
Naxos disease JUP Plakoglobin 17q21 AR
ARVD: arrhythmogenic right ventricular dysplasia, AD: autosomal dominant, TGFB: transforming growth factor beta, RYR: ryanodine receptor, TMEM: transmembrane protein, DSP: desmoplakin, AR: autosomal recessive, PKP: plakophilin, DSG: desmoglein, JUP: junction plakoglobin
Fig. 4. Endomyocardial biopsy from patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia showing typical fatty (A) and fibrofatty (B) infiltration.
A B
520·Arrhythmogenic RV Cardiomyopathy/Dysplasia
American study, VT induction was associated with an increased risk for ICD therapy in patients with ARVC/
D.48) Different populations, different types of interven- tions, different ventricular arrhythmias requiring ICD intervention, and a relatively high rate of ICD therapy in the American trial may explain the differences.
Differential Diagnosis
Some patients with localized ARVC/D, particularly in the infundibulum, may exhibit normal RV volumes and a preserved RV ejection fraction, and may present clinically as idiopathic RV outflow tract tachycardia.49) However, inducibility of VT, presence of more than one ECG morphology, and fragmented potentials may help- ful in distinguishing ARVC/D-associated VT from idi- opathic RV outflow tract tachycardia.31) It is important to determine whether the patient has idiopathic VT or ARVC/D for two reasons. First, idiopathic VT is a benign condition and ICD implantation is usually not recom- mended. Second, because ARVC/D is a hereditary dis- ease, screening all first-degree family members is recom- mended.11) Some definite cases of biopsy-proven myo- carditis showed typical clinical findings of ARVC/ D.46)
Risk Stratification
The available data suggest that young age, prior car- diac arrest, fast and poorly tolerated VT with different morphologies, syncope, severe RV dysfunction, heart failure with LV involvement, and familial occurrence of juvenile sudden death are the major determinants in predicting sudden cardiac death and clinical outcomes with a poor prognosis.5)29)40)48)50-52)
Even the presence of sustained VT in patients with heart failure is an important risk factor.29) Appropriate ICD therapy during follow-up is also observed fre- quently in young patients considered to be at high risk with previous cardiac arrest, VT with hemodynamic com- promise, and LV involvement.40)
Management
There is no curative treatment for ARVC/D; rather, the aim is to detect patients at high risk and prevent complications. The four therapeutic options for ARVC/
D include antiarrhythmic drugs, catheter ablation, ICD implantation, and surgery. Once the diagnosis has been established, treatment should include avoiding compe- titive sports or vigorous exertion, initiation of a beta- blocker, and consideration for ICD placement.33) ICDs are recommended for all patients with ARVC/D who have experienced aborted sudden cardiac death or a sus- tained ventricular arrhythmia. Catheter ablation should be considered in patients with recurrent episodes of VT,
despite antiarrhythmic therapy, but should not be con- sidered to be an alternative to ICD therapy.11) When the disease has progressed to RV or biventricular failure, treatment consists of the current therapy for heart fa- ilure, including diuretics, beta-blocking agents, angio- tensin-converting enzyme inhibitors, and anticoagula- tion. In case of intractable RV failure, cardiac transplan- tation may be the only remaining alternative.31)53)
Antiarrhythmic Agents
In patients with ARVC/D at low risk for arrhythmic sudden death, specific antiarrhythmic treatment is usually not required. However, should a patient still suffer severe symptoms from palpitations, treatment with con- ventional antiarrhythmic drugs may be considered. Spe- cific antiarrhythmic drugs or catheter ablation should be limited to patients with significant symptoms refrac- tory to these measures.52) Sotalol was shown to be the most effective antiarrhythmic drug to prevent inducible VT in patients with ARVC/D in a German study.54) In the subgroup of high-risk patients, ICD implantation is life-saving and results in improved survival compared with pharmacologic therapy, but antiarrhythmic drugs are often administered to reduce the need for cardiover- sion or inappropriate interventions.40)48)51)
Catheter Ablation
Catheter ablation is considered a palliative procedure in patients suffering from a high frequency of ICD dis- charges due to recurrent monomorphic VT.52) Because of the progressive nature of the disease, catheter ablation is not considered to be a curative procedure.11) Previous studies have shown a favorable acute success rate with catheter ablation of RV tachycardia.37)55) However, in pa- tients with ARVC/D, VT recurrences during follow-up are common. The discrepancy between the favorable acute results and the unfavorable long-term outcome may be explained by the progressive nature of ARVC/D, which predisposes to the occurrence of new and malignant ar- rhythmogenic substrates over time.49)
Implantable Cardioverter-Defibrillator
ICDs are recommended for all patients with ARVC/
D who have experienced aborted cardiac death or a sus- tained ventricular arrhythmia.11) However, no prospective randomized trials have compared ICD implantation with other treatment modalities in patients with ARVC/D.
ICD therapy is safe and effective in patients with ARVC/D and provides lifesaving protection.40)48)
Due to the structural abnormalities of the RV myo- cardium in patients with ARVC/D, meticulous atten- tion has to be paid to the placement of the RV defibril-
Yongkeun Cho·521
lation lead during lead implantation, in order to achieve satisfactory acute and long-term pacing and sensing results. Progression of myocardial atrophy and subse- quent replacement by fat and fibrosis at the site of lead implantation may result in a loss of sensing function of the RV defibrillation lead and may require lead revision or the implantation of an additional pace/sense lead.40)51)
Treatment of Heart Failure
Symptomatic heart failure requiring treatment is present in only 10-20% of patients with ARVC/D.
Symptomatic heart failure is unusual as an early mani- festation of ARVC/D and occurs almost exclusively in patients with an advanced stage of the disease. In addi- tion to progressive dysfunction of the RV, these patients frequently demonstrate LV involvement, and therefore exhibit clinical symptoms of biventricular heart failure.
With the lack of causal treatment options for heart failure in ARVC/D, conventional pharmacologic therapy, including vasodilators, diuretics, beta-blockers, and di- gitalis is required. Some of the patients have undergone heart transplantation for intractable heart failure asso- ciated with ventricular tachyarrhythmias or a marked increase in the intracardiac defibrillation threshold in the presence of repeated episodes of VT.5)48)51)52)
Management of Asymptomatic Patients With Family Members
Asymptomatic patients with ARVC/D do not require specific treatment. However, they should be followed by regular non-invasive cardiac testing for the early detec- tion of ventricular arrhythmias and the potential pro- gression of the disease.13) Twelve-lead ECG and 2-D echocardiography represent essential baseline diagnostic investigations. Exercise testing, Holter monitoring, and signal-averaged ECG may be added to the previous es- sential studies. If the results of such testing reveals sus- picious ARVC/D or complex ventricular arrhythmias, more detailed studies should be performed.52) Among the relatives of probands with ARVC/D, a minority ful- fills diagnostic criteria for dilated cardiomyopathy with-
out any evidence of RV disease.27)
Exercise and ARVC/D
ARVC/D is a frequent cause of sudden cardiac death in athletes.25) Patients with ARVC/D should be advised against participation in competitive sports, since this appears to be associated with accelerated disease progres- sion and an increased risk of sudden death.34)49)52)53)56) Ac- tivities, such as golf or walking, are encouraged, whereas activities, such as marathon running or weight lifting, should be strongly discouraged.11) Sports activities, par- ticularly running and bicycling, facilitate disruption of the myocardial cells at an earlier age.34) Recently, endu- rance training-induced RV enlargement has been re- ported in animal models.56)
Overlap Between Brugada Syndrome and ARVC/D
Among patients with ARVC/D, there is a subpopula- tion that exhibits a clinical and ECG pattern similar to that of patients with the Brugada syndrome.33)57)58) En- domyocardial biopsy showed typical fibrofatty infiltration of the myocardium in some patients with ECG features typical of the Brugada syndrome.57) Even ajmaline chal- lenge showed positive results in some of the cases.58) Those cases are thought to represent an early or con- cealed form of the disease.
Prognosis
Long-term follow-up studies in large populations are not yet available, making the prognosis in symptomatic patients difficult to define. There is even less informa- tion about the clinical outcome of asymptomatic, affect- ed individuals. The limited data on risk stratification indicate that patients with severe RV dysfunction, LV involvement, a history of syncope or cardiac arrest, family history, and inducible VT are more prone to life-threat- ening VT and sudden death (Table 4). However, no pro- spective or randomized studies have investigated the com- parative efficacy of these different treatment options
Table 4. Proposed risk factors for unfavorable prognosis in arrhythmogenic right ventricular cardiomyopathy/dysplasia39)
Clinical marker Assessment
Previous cardiac arrest* Clinical history Syncope or sustained ventricular tachycardia
with impairment of consciousness*
Direct inquiry regarding symptoms
Increased QRS dispersion A difference of ≥40 ms between the maximum and minimum QRS values occurring in any of the 12 ECG leads
Early onset of symptoms Direct inquiry regarding symptoms
Severe right ventricular dilation Echocardiography or cardiac magnetic resonance
Left ventricular involvement* Regional wall motion abnormalities or dilation and impairment of systolic function in the left ventricle inverted T waves in the precordial leads beyond V3
*clinical markers that warrant consideration of implantable cardioverter-defibrillator therapy
522·Arrhythmogenic RV Cardiomyopathy/Dysplasia
thus far.52)
As ARVC/D is a progressive disease that involves the RV, partially or globally, and becoming more diffusely damaged with time, the existence of RV failure implies progression and severity of the disease. Although ma- croscopic or histologic changes of the LV are found in many patients, LV failure is uncommon and it is present only in later stages of the disease. The biventricular fail- ure mimics a dilated cardiomyopathy.31)
Conclusions
ARVC/D is a type of cardiomyopathy characterized by RV involvement and the risk of arrhythmic sudden death. Distinctive pathologic features are ventricular myo- cardial atrophy with replacement by fatty or fibrofatty tissue. Patients affected with ARVC/D should be exclud- ed from competitive sports and vigorous training. An- tiarrhythmic drugs may be used to suppress ventricular arrhythmias in patients with a low risk of sudden death.
Catheter ablation shows favorable acute results for eli- minating ventricular arrhythmias. However, recurrent arrhythmias from new foci are frequent during follow- up. ICD therapy has been increasingly used for secondary and primary prevention of sudden death. Greater aware- ness and understanding of ARVC/D among physicians may prevent unnecessary deaths, especially in the young.
REFERENCES
1) Marcus FI, Fontaine GH, Guiraudon G, et al. Right ventricular dysplasia: a report of 24 adult cases. Circulation 1982;65:384-98.
2) Thiene G, Nava A, Corrado D, Rossi L, Pennelli N. Right ven- tricular cardiomyopathy and sudden death in young people. N Engl J Med 1988;318:129-33.
3) Choi KJ, Kwon HC, Nam CB, et al. A case of arrhythmogenic right ventricular dysplasia. Korean Circ J 1995;25:1057-63.
4) Kim YK, Han DS, Kweon SH, Lee MI, Lee HJ. A case of arrhythmogenic right ventricular dysplasia. Korean Circ J 1996;
26:1204-9.
5) Corrado D, Basso C, Thiene G, et al. Spectrum of clinicopa- thologic manifestations of arrhythmogenic right ventricular car- diomyopathy/dysplasia: a multicenter study. J Am Coll Cardiol 1997;30:1512-20.
6) Park TI, Kim DJ, Sohn YK, et al. Arrhythmogenic right ventri- cular cardiomyopathy as a cause of sudden unexplained death.
Korean Circ J 2001;31:335-40.
7) Cho Y, Park T, Yang DH, et al. Arrhythmogenic right ventricular cardiomyopathy and sudden cardiac death in young Koreans.
Circ J 2003;67:925-8.
8) Cho Y, Park T, Shin D, et al. Clinical manifestations of arrhyth- mogenic right ventricular cardiomyopathy in Korean patients.
Int J Cardiol 2007;122:137-42.
9) Fox PR, Maron BJ, Basso C, Liu SK, Thiene G. Spontaneously occurring arrhythmogenic right ventricular cardiomyopathy in the domestic cat: a new animal model similar to the human disease. Circulation 2000;102:1863-70.
10) Basso C, Fox PR, Meurs KM, et al. Arrhythmogenic right ven- tricular cardiomyopathy causing sudden cardiac death in boxer
dogs: a new animal model of human disease. Circulation 2004;
109:1180-5.
11) Calkins H. Arrhythmogenic right-ventricular dysplasia/cardio- myopathy. Curr Opin Cardiol 2006;21:55-63.
12) Nava A, Thiene G, Canciani B, et al. Familial occurrence of right ventricular dysplasia: a study involving nine families. J Am Coll Cardiol 1988;12:1222-8.
13) Nava A, Bauce B, Basso C, et al. Clinical profile and long-term follow-up of 37 families with arrhythmogenic right ventricular cardiomyopathy. J Am Coll Cardiol 2000;36:2226-33.
14) Coonar AS, Protonotarios N, Tsatsopoulou A, et al. Gene for arrhythmogenic right ventricular cardiomyopathy with diffuse nonepidermolytic palmoplantar keratoderma and woolly hair (Naxos disease) maps to 17q21. Circulation 1998;97:2049-58.
15) McKoy G, Protonotarios N, Crosby A, et al. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular car- diomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease). Lancet 2000;355:2119-24.
16) Alcalai R, Metzger S, Rosenheck S, Meiner V, Chajek-Shaul T.
A recessive mutation in desmoplakin causes arrhythmogenic right ventricular dysplasia, skin disorder, and woolly hair. J Am Coll Cardiol 2003;42:319-27.
17) Rampazzo A, Nava A, Danieli GA, et al. The gene for arrhyth- mogenic right ventricular cardiomyopathy maps to chromosome 14q23-q24. Hum Mol Genet 1994;3:959-62.
18) Rampazzo A, Nava A, Erne P, et al. A new locus for arrhythmo- genic right ventricular cardiomyopathy (ARVD2) maps to ch- romosome 1q42-q43. Hum Mol Genet 1995;4:2151-4.
19) Severini GM, Krajinovic M, Pinamonti B, et al. A new locus for arrhythmogenic right ventricular dysplasia on the long arm of chromosome 14. Genomics 1996;31:193-200.
20) Rampazzo A, Nava A, Miorin M, et al. ARVD4, a new locus for arrhythmogenic right ventricular cardiomyopathy, maps to chro- mosome 2 long arm. Genomics 1997;45:259-63.
21) Ahmad F, Li D, Karibe A, et al. Localization of a gene respon- sible for arrhythmogenic right ventricular dysplasia to chromo- some 3p23. Circulation 1998;98:2791-5.
22) Li D, Ahmad F, Gardner MJ, et al. The locus of a novel gene responsible for arrhythmogenic right ventricular dysplasia char- acterized by early onset and high penetrance maps to chromo- some 10p12-p14. Am J Hum Genet 2000;66:148-56.
23) Mallat Z, Tedgui A, Fontaliran F, Frank R, Durigon M, Fontaine G. Evidence of apoptosis in arrhythmogenic right ventricular dys- plasia. N Engl J Med 1996;335:1190-6.
24) Valente M, Calabrese F, Thiene G, et al. In vivo evidence of ap- optosis in arrhythmogenic right ventricular cardiomyopathy. Am J Pathol 1998;152:479-84.
25) Corrado D, Thiene G, Nava A, Rossi L, Pennelli N. Sudden death in young competitive athletes: clinicopathologic correla- tions in 22 cases. Am J Med 1990;89:588-96.
26) Hermida JS, Minassian A, Jarry G, et al. Familial incidence of late ventricular potentials and electrocardiographic abnormalities in arrhythmogenic right ventricular dysplasia. Am J Cardiol 1997;79:1375-80.
27) Hamid MS, Norman M, Quraishi A, et al. Prospective evaluation of relatives for familial arrhythmogenic right ventricular car- diomyopathy/dysplasia reveals a need to broaden diagnostic cri- teria. J Am Coll Cardiol 2002;40:1445-50.
28) Corrado D, Basso C, Thiene G. Arrhythmogenic right ventricular cardiomyopathy: diagnosis, prognosis, and treatment. Heart 2000;
83:588-95.
29) Hulot JS, Jouven X, Empana JP, Frank R, Fontaine G. Natural history and risk stratification of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circulation 2004;110:1879-84.
Yongkeun Cho·523
30) Burke AP, Farb A, Tashko G, Viramini R. Arrhythmogenic right ventricular cardiomyopathy and fatty replacement of the right ventricular myocardium: are they different disease? Circulation 1998;97:1571-80.
31) Kies P, Bootsma M, Bax J, Schalij MJ, van der Wall EE. Ar- rhythmogenic right ventricular dysplasia/cardiomyopathy: screen- ing, diagnosis, and treatment. Heart Rhythm 2006;3:225-34.
32) Tabib A, Loire R, Chalabreysse L, et al. Circumstances of death and gross and microscopic observations in a series of 200 cases of sudden death associated with arrhythmogenic right ventricular cardiomyopathy and/or dysplasia. Circulation 2003;108:3000-5.
33) Frances RJ. Arrhythmogenic right ventricular dysplasia/cardio- myopathy: a review and update. Int J Cardiol 2006;110:279-87.
34) Heidbüchel H, Hoogsteen J, Fagard R, et al. High prevalence of right ventricular involvement in endurance athletes with ventri- cular arrhythmias: role of an electrophysiologic study in risk stratification. Eur Heart J 2003;24:1473-80.
35) McKenna WJ, Thiene G, Nava A, et al. Diagnosis of arrhythmo- genic right ventricular dysplasia/cardiomyopathy. Br Heart J 1994;71:215-8.
36) Bomma C, Rutberg J, Tandri H, et al. Misdiagnosis of arrhythmo- genic right ventricular dysplasia/cardiomyopathy. J Cardiovasc Electrophysiol 2004;15:300-6.
37) Marchlinski FE, Zado E, Dixit S, et al. Electroanatomic substrate and outcome of catheter ablative therapy for ventricular tachy- cardia in setting of right ventricular cardiomyopathy. Circula- tion 2004;110:2293-8.
38) Nasir K, Bomma C, Tandri H, et al. Electrocardiographic features of arrhythmogenic right ventricular dysplasia/cardiomyopathy according to disease severity: a need to broaden diagnostic cri- teria. Circulation 2004;110:1527-34.
39) Sen-Chowdhry S, Lowe MD, Sporton SC, McKenna WJ. Ar- rhythmogenic right ventricular cardiomyopathy: clinical presen- tation, diagnosis, and management. Am J Med 2004;117:685-95.
40) Corrado D, Leoni L, Link MS, et al. Implantable cardioverter- defibrillator therapy for prevention of sudden death in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia.
Circulation 2003;108:3084-91.
41) Tonet JL, Castro-Miranda R, Iwa T, Poulain F, Frank R, Fontaine GH. Frequency of supraventricular tachyarrhythmias in arrhy- thmogenic right ventricular dysplasia. Am J Cardiol 1991;67:
1153.
42) Yoerger DM, Marcus F, Sherill D, et al. Echocardiographic findings in patients meeting task force criteria for arrhythmo- genic right ventricular dysplasia: new insights from the multi- disciplinary study of right ventricular dysplasia. J Am Coll Cardiol 2005;45:860-5.
43) Tandri H, Saranathan M, Rodriguez ER, et al. Noninvasive detection of myocardial fibrosis in arrhythmogenic right ventri- cular cardiomyopathy using delayed-enhancement magnetic re- sonance imaging. J Am Coll Cardiol 2005;45:98-103.
44) Sen-Chowdhry S, Prasad SK, Syrris P, et al. Cardiovascular mag- netic resonance in arrhythmogenic right ventricular cardiomyo-
pathy revisited: comparison with task force criteria and geno- type. J Am Coll Cardiol 2006;48:2132-40.
45) Bomma C, Dalal D, Tandri H, et al. Evolving role of multide- tector computed tomography in evaluation of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Am J Cardiol 2007;
100:99-105.
46) Chimenti C, Pieroni M, Maseri A, Frustaci A. Histologic find- ings in patients with clinical and instrumental diagnosis of sporadic arrhythmogenic right ventricular dysplasia. J Am Coll Cardiol 2004;43:2305-13.
47) Moric-Janiszewska E, Markiewicz-Loskot G. Review on the genetics of arrhythmogenic right ventricular dysplasia. Europace 2007;9:259-66.
48) Roguin A, Bomma CS, Nasir K, et al. Implantable cardioverter- defibrillators in patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy. J Am Coll Cardiol 2004;43:1843-52.
49) Corrado D, Basso C, Leoni L, et al. Three-dimensional electroa- natomical voltage mapping and histologic evaluation of myocar- dial substrate in right ventricular outflow tract tachycardia. J Am Coll Cardiol 2008;51:731-9.
50) Peters S, Peters H, Thierfelder L. Risk stratification of sudden cardiac death and malignant ventricular arrhythmias in right ventricular dysplasia-cardiomyopathy. Int J Cardiol 1999;71:
243-50.
51) Wichetr T, Paul M, Wollmann C, et al. Implantable cardioverter/
defibrillator therapy in arrhythmogenic right ventricular car- diomyopathy: single-center experience of long-term follow-up and complications in 60 patients. Circulation 2004;109:1503-8.
52) Wichter T, Paul M, Eckardt L, et al. Arrhythmogenic right ventricular cardiomyopathy: antiarrhythmic drugs, catheter abla- tion, or ICD? Herz 2005;30:91-101.
53) Buja G, Estes NA 3rd, Wichter T, Corrado D, Marcus F, Thiene G. Arrhythmogenic right ventricular cardiomyopathy/dysplasia:
risk stratification and therapy. Prog Cardiovasc Dis 2008;50:
282-93.
54) Wichter T, Borggrefe M, Haverkamp W, Chen X, Breithardt G.
Efficacy of antiarrhythmic drugs in patients with arrhythmogenic right ventricular disease: results in patients with inducible and noninducible ventricular tachycardia. Circulation 1992;86:29-37.
55) Verma A, Kilicaslan F, Schweikert RA, et al. Short- and long- term success of substrate-based mapping and ablation of ventri- cular tachycardia in arrhythmogenic right ventricular dysplasia.
Circulation 2005;111:3209-16.
56) Kirchhof P, Fabritz L, Zwiener M, et al. Age- and training- dependent development of arrhythmogenic right ventricular car- diomyopathy in heterozygous plakoglobin-deficient mice. Cir- culation 2006;114:1799-806.
57) Kim H, Cho Y, Park Y, et al. Underlying cardiomyopathy in pa- tients with ST-segment elevation in the right precordial leads.
Circ J 2006;70:719-25.
58) Peters S, Trümmel M, Denecke S, Koehler B. Results of ajmaline testing in patients with arrhythmogenic right ventricular dysplasia- cardiomyopathy. Int J Cardiol 2004;95:207-10.