총 설
제올라이트와 분자체 촉매에서 메탄올 전환 반응의 기구
서 곤†·민병구 전남대학교응용화학공학부
500-757 광주시북구용봉동 300 (2006년 7월 18일접수, 2006년 7월 26일채택)
Mechanism of Methanol Conversion over Zeolite and Molecular Sieve Catalysts
Gon Seo† and Byung Goo Min
School of Applied Chemical Engineering, Chonnam National University, 300, Yongbong-dong, Puk-gu, Gwangju 500-757, Korea (Received 18 July 2006; accepted 26 July 2006)
요 약
원유가급등으로메탄올에서저급올레핀을제조하는공정에대한관심이높아지고있다. 제올라이트와 SAPO 분자 체촉매에서메탄올의탄화수소로전환반응을저급올레핀생성단계에중점을두고고찰하였다. 구조가명확한중간 체를근거로하는직접(direct) 반응기구와구조가애매한탄화수소뭉치를활성점으로보는탄화수소활성체(hydrocarbon pool) 반응기구의합리성을, 메탄올전환반응의유도기간, 생성물의선택성, 활성저하등과연계지어비교하였다. 탄 화수소활성체의구조규명과메탄올전환반응에서촉매활성점으로서기능에대한 1999년이후연구결과를검토하 였으며, 메탄올에서저급올레핀을생산하는공정에대한전망도기술하였다.
Abstract −The production of lower olefins from methanol becomes an attractive process because of the rapid increase in crude oilprice. This paper reivews the conversion mechanisms of methanol to hydrocarbons over zeolite and SAPO molecular sieve catalysts to understand the formation steps of lower olefins from methanol. The feasibility of the con- version mechanisms such as the direct mechanism based on well-defined intermediates and the hydrocarbon pool mech- anism involving hydrocarbon moieties as an active centers is discussed with reepect to the induction period, the selectivity for products and the deactivation phenomena of the methanol conversion. The literature appeered since 1999 for the structure of the hydrocarbon pool and its catalytic role in the methanol conversion are summariged, and the pros- pect for the methanol-to-olefins process is described.
Key words: Methanol, Lower Olefins, MTO, MFI Zeolite, SAPO Molecular Sieve
1. 서 론
석탄이나천연가스에서만든메탄올을한단계반응을거쳐고급 가솔린으로전환시킬수있다는모빌(Mobil)사의메탄올의가솔린 으로전환(methanol to gasoline: MTG) 공정은오일쇼크에서벗어
나지못하고있던 1970년대상황에서아주놀라운연구결과이었다
[1]. 다양한탄화수소자원에서제조한합성가스로부터메탄올을합
성하고, 이로부터한단계를거쳐가솔린을제조하므로, 원유에전 적으로의존하던연료유나석유화학공업의원료를다양화할수있 는획기적인성과로인정받았다. 촉매의성질과반응조건을적절 히조절하면메탄올로부터가솔린외에저급올레핀, 디젤유, 방향 족화합물등을선택적으로제조할수있다는사실이알려지면서[2, 3],
메탄올의탄화수소로전환공정에대한연구는매우활발하게진
행되었다[4]. 뿐만아니라이 공정의촉매인 MFI 제올라이트와
SAPO-34 분자체에대한관심도높았다[5]. 수요에맞추어목적생
성물을탄력적으로생산할수있다는점에서, 메탄올을중간물질
로설정하는석유화학산업의새로운체계가거론될만큼 MTG 공
정의파장이컸다.
이러한기대와달리 MTG 공정은산업으로는성장하지못했다.
원유가격이급락하면서천연가스에서메탄올을거쳐가솔린을제 조하는공정이경쟁력을잃었다. 자국의가솔린수요일부를스스
로생산하기위한정치적목적으로 1985년뉴질랜드에년산 70만
톤규모의가솔린을생산하는 MTG 공장이건설되었으나, 더이상
의산업화실적은없다[4]. 오일쇼크이후예상과달리원유가격
이오랫동안낮게유지되면서 MTG 공정뿐아니라원유이외의대 체자원에서가솔린을제조하려던대부분의시도가중단되었다. 뉴
질랜드의 MTG 공장조차도현재는합성가스생산공정등공장시
설의일부만을운용하고있다.
†To whom correspondence should be addressed.
E-mail: [email protected]
MTG 공정으로생산한가솔린의가격경쟁력은원료인천연가스 의가격과경쟁공정의원료인원유가격에의해결정된다. 합성가 스를생산하고이로부터메탄올을거쳐가솔린을제조하는공정은 단계별로압력변화가커서시설비부담이크므로, 가솔린가격이 생산규모에따라어느정도달라진다. 그러나가솔린제조단계를 제외한합성가스의제조나메탄올합성공정은역사가길고잘정 립된기술이어서, 공정기술의차이는그리크지않으므로원료가 격이공정경쟁력의가장중요한인자가된다. 연구자에따라수치 에차이가있지만, 천연가스의가격이아주낮게잡아도원유가격 이배럴당 35달러이상이어야 MTG 공정이가격경쟁력을가진다
는전망이설득력이있다[3]. 따라서원유가격이배럴당 20달러정
도로낮게유지되던시절에는 MTG 공정의상업화는가격경쟁력
면에서불가능한일이었다.
MTG 공정은산업으로는성공하지못했지만이공정의촉매인
MFI 제올라이트는이후꾸준히연구되었다. MFI 제올라이트는처 음합성한모빌사의옛이름(Socony Mobil)을따서 ZSM-5 제올라
이트라고부르기도한다. Fig. 1에보인것처럼 MFI 제올라이트의
세공구조는매우특이하고, 골격은오각형(pentasil) 구조로이루어
져있다. 10개산소원자고리로이루어진선형(straight) 세공과구 부러진(sinusoidal) 세공이교차하는독특한세공구조, Si/Al 몰비를
10 근처에서부터무한대까지바꿀수있는매우가변적인산성도,
골격의우수한수열안정성, 형상선택성이나타나는세공크기등
이 MFI 제올라이트의특징이다. 구조가안정하고 Si/Al 몰비의조
절이용이하여산성도를폭넓게변화시킬수있고, 후처리나담지 등으로촉매성질을바꿀수있어서촉매재료로서가능성이많다.
인담지로인한산성도조절, 실리카담지로인한세공크기조절,
골격에철이나갈륨등다양한원소의치환등 MFI 제올라이트의
촉매성질을조절하는방안도많이연구되었다.
MFI 제올라이트는규칙적인세공구조를가진물질에서나타나 는형상선택성이가장뚜렷하게나타나는제올라이트로서파라-
선택성이 우수하여 p-자일렌을 제조하는 자일렌의 이성질화
(isomerization) 공정과톨루엔의알킬화(alkylation) 공정에촉매로
사용한다. 이외에도왁스제거(dewaxing) 공정과 LPG에서방향족
화합물을제조하는 CYCLAR 공정에촉매로활용된다[6]. 기대하는
생성물에대한선택성을높이기위해 FCC 공정의촉매에도일정량
첨가된다. 탄소침적에의한활성저하가느려고분자물질의분해 반응에서도촉매활성이매우우수하다[7].
최근원유가격이급등하여배럴당 70달러를넘어섰다. 예측이기 는하지만, 배럴당 100달러를넘어설가능성도제기되고있다. 원유 를원료로사용하는정유공업과석유화학공업의붕괴위험성이 거론되면서, 원유이외의대체물질에서연료유와석유화학공업의
원료를확보해야할필요성이절실해지고있다. 바이오에탄올과바 이오디젤의보급이활발하게추진되고있으나, 주로차량의연료유 대체에국한되고있다. 석유화학공업에서도석탄이나천연가스를 원유의대체원료로고려하고있다. 특히대체원료에서제조한메 탄올로부터석유화학공업의원료인저급올레핀을제조하는공정 에관심이많다.
최근생산규모가백만톤이상인대형메탄올제조공장이운전 되면서메탄올가격이낮아지고안정적인공급을기대할수있게 되었다. 원유가급등에비해메탄올의안정적공급은메탄올에서저 급올레핀을제조하는공정의경쟁력을높여주고있다. 나프타를열 분해하여저급올레핀을제조하는열분해공정은원유에서증류하 여제조하는나프타가격이비싸져서가격경쟁력이약해지고, 과 다한에너지사용으로온실가스인이산화탄소를많이배출한다는 환경부담도크다. 이런점에서메탄올에서저급올레핀을생산
(methanol to olefin: MTO)하는공정은나프타열분해공정을대체 하지는못하더라도보완할수있는공정으로서의미가커지고있다.
MTO 공정은꾸준히연구개발되어산업화가가능한수준에이
른걸로이야기되고있다. MFI 제올라이트를사용하는모빌사의
MTO 공정이나 SAPO-34 분자체를사용하는유니온카바이드
(Union Carbide)사의공정은상업화수준까지개발되어있다[4]. 중 국의대련화학물리연구소에서도 SAPO-34 분자체계촉매를사용 한메탄올에서저급올레핀생산공정의연구를파일럿공장수준
까지운전한경험이있다[8]. 이런점에서가격경쟁력만확보된다
면 MTO 공정의상용공장이바로건설되는데기술적인문제는없
다고판단된다.
메탄올에서올레핀등탄화수소를제조하는공정의반응기구연 구도같이이루어졌다. 메탄올전환반응은메탄올→디메틸에테르
(dimethyl ether)→저급올레핀→파라핀과방향족탄화수소로반응
의진행경로가확실하고, 반응조건이나촉매성질에따라생성물
분포가크게달라진다[1, 2]. 그러나메탄올에서탄화수소가생성되
는반응기구에대해서는이견이많다. 반응은매우단순해보이지 만, 20여가지반응기구가제시될정도로반응기구에대한논란이 있다.
제안된반응기구는크게카벤, 옥소니움이온, 카르보니움이온,
자유라디칼등특정한구조의중간체를거치는반응기구와이와달 리구조를확실히알수없는중간체를가정한반응기구로나눌수 있다[9]. 구조가명확한중간체(distinctive intermediate)를가정한반
응기구(direct mechanism)로는중간체에서탄화수소가바로생성되
므로탄소-탄소결합이생성될때나타나는유도기간과생성물중 프로필렌과에틸렌의선택성, 제올라이트의세공구조가생성물분 포에미치는영향을설명하기어렵다. 이에비해구조가애매한중 간체(indistinctive intermediate)를가정한반응기구에서는중간체자 체가확실하지않기때문에이들의역할이나중간체에서탄화수소 가생성되는단계를설득력있게설명하지못한다. 그러나중간체 인탄화수소뭉치가생성되는데필요한시간으로유도기간을, 탄화 수소뭉치의구조와생성물의선택성을서로연관지을수있다. 최 근분석기기와측정기술의발달로촉매내에형성된탄화수소의 구조와상태가규명되어중간체의구조가알려지면서실험결과나 현상설명에적합한반응기구로주목받고있다. 탄화수소임은확실
하지만, 구체적인구조를몰라탄소나탄화수소뭉치(carbon or
hydrocarbon pool)라고불렀던중간체의구조가실험적으로밝혀지
Fig. 1. Pore structure and skeletal of MFI zeolite.
면서, 이제는설득력있는반응기구로인정받고있다.
이러한반응기구에대한연구성과는극소량의생성물을검출할
수있으면서생성물내동위원소분포를파악할수있는 GC-MS가
연결된반응기와촉매내에형성된탄화수소의구조와상태를직접 파악할수있는동시조작(in situ) 고체 NMR의효과적인연계활용
의결실이다[10, 11]. 반응도중촉매에축적되는물질과생성물분
포를근거로가설수준이던탄화수소뭉치가 Kolboe와 Haw의연
구팀에의해촉매활성점으로구체화되었다[12-21]. 초기에는침적 탄소(coke)와유사하다고생각하여 ‘탄소뭉치(carbon pool)’이라고 불렀으나, 구조가밝혀지면서탄화수소라고부르는게적당하다고 하여 ‘hydrocarbon pool’로이름이바뀌었다. 영어의 ‘pool’을그대 로번역하면 ‘덩어리’나 ‘뭉치’가적절하므로 ‘hydrocarbon pool
mechanism’을 ‘탄화수소뭉치반응기구’로써야하겠지만탄화수소
뭉치가전환반응의활성점으로작용한다는점에서 ‘활성체’가실 제기능을반영한다는점에서더적절하다. 외국어를그대로옮기 는것보다우리말로의미를떠올릴수있는용어가더적절하다는 뜻에서이총설에서는 ‘hydrocarbon pool mechanism’을 ‘탄화수소 활성체반응기구’로부르기로한다. 세공내에생성된탄화수소활 성체를중간체로설정하므로그간제기되었던메탄올전환반응의 유도기간이나생성물선택성에대한의문이상당부분해소되었고,
메탄올에서저급올레핀이생성되는단계를구체적으로설명할수 있다.
원유자원이거의없어석유화학산업의원료를대부분수입에 의존하고있는우리나라에서는저급올레핀의수급상황이석유화 학산업의경쟁력에미치는영향이매우크다. 이런현실을감안하 여원유가격의급등에대비하고원료를원활하게확보하는데메탄
올에서저급올레핀을제조하는 MTO 공정이대안이될수있다.
나아가새로운촉매의개발이나효율적인공정구성도모색해볼 수있다. 이총설에서는이러한 MTO 공정의연구개발에도움이 되었으면하는뜻으로메탄올의탄화수소로전환반응에서반응기 구에대한최근의연구결과를종합정리하였다. MTO 공정자체에
대해서는 1999년에발간된 Keil의총설이있어다루지않았다[4].
이문헌에서는 MTO뿐아니라 MTG 공정도다루고있으며, 액상에 서디메틸에테르를제조하는공정도소개하고있다. 촉매에대해서 는 Stöcker의총설이있다[5]. 촉매의입자크기, 세공구조, 산성도,
반응조건등이촉매활성과선택성에미치는영향을잘기술하고 있다. 메탄올전환반응에관련된반응기설계, 확산현상, 속도론 적고찰, 반응물조성의영향이나탄화수소분해반응과복합운전
등에관한연구도많으나[22-30], 반응기구와연관성이작아거론하
지않았다. 제올라이트나분자체세공내에서탄화수소활성체가어 떻게형성되고, 메탄올전환반응에서이들이촉매로어떻게작용 하는가하는데에중점을두었다.
2. 메탄올의 탄화수소로 전환 반응
메탄올에서탄화수소가생성되는반응에대해서는 Chang의총설
[1, 2]에잘정리되어있으므로, 자세히언급하지않는다. 1880년기 록에도메탄올에서탄화수소의합성이언급될만큼이전환반응에 대한관심은많으나, 메탄올에서탄화수소의합성은그리쉽지않 았다. 알루미나를촉매로사용하면메탄올이디메틸에테르로정량 적으로전환되나, 이들이모두탄화수소로전환된다는보고는없다.
소량의탄화수소가부산물로생성되기는해도주생성물이라고볼
수있을만큼전환되지않는다. 1950년대에도메탄올에서탄화수소
왁스를제조하는특허가출원되었다는점에서, 메탄올에서탄화수 소를합성하려는시도는계속되었다고생각한다. 그러나메탄올을 제조하여탄화수소로합성하는공정보다는합성가스에서바로
Fischer-Tropsch 반응을거쳐탄화수소를생산하는공정이더빨리
상업화되었다. 이공정역시원유에서탄화수소를생산하는정유공 정에밀려사라졌다. 다만, 인종차별로석유구입에어려움을겪던 남아프리카공화국에서 SASOL 공정이라는이름으로지금도운전되 고있다.
MFI 제올라이트를촉매로사용하면메탄올은탄화수소로쉽게
전환된다. Fig. 2에 MFI 제올라이트촉매에서접촉시간에따른메
탄올전환반응의생성물분포를보였다[1]. 메탄올이탈수되어디
메틸에테르가생성되고, 이로부터저급올레핀이생성된다. 저급올 레핀은다시파라핀과방향족화합물로전환되므로, 메탄올이탄화 수소로전환되는반응의경로를다음처럼요약할수있다.
올레핀에서파라핀이생성되려면수소가추가로공급되어야하나,
방향족화합물이생성될때수소가방출되기때문에파라핀과방향 족화합물이같이생성되면탄소침적을고려하지않아도물질수 지가성립한다. 메탄올이탄화수소로전환되는반응의생성물분포
는반응온도에따라서도상당히다르다[1, 2]. Fig. 3에서보듯이
아주낮은온도에서는디메틸에테르가주생성물이나온도가높아지 면 C2~C5탄화수소가많이생성된다. 온도가더높아지면방향족 화합물의분율이높아지나, 아주높아지면메탄과올레핀함량이많 아진다. 낮은온도에서는디메틸에테르를거쳐저급올레핀이생성 되는단계까지만반응이진행되지만, 온도가높아지면방향족화합 물이많이생성된다. 이와함께온도가높아지면분해생성물이많 아져서메탄이나일산화탄소등이생성물에서검출된다. 생성물분 포는접촉시간과온도외에도반응계의압력에따라서도달라진다.
개략적이긴하지만 Fig. 2와 3 결과는앞에정리한반응경로의타 당성을잘보여주고있다.
12개산소원자고리로이루어진큰세공(large pore)이있는 FAU Fig. 2. Variation of product composition with space velocity in the
methanol conversion over MFI zeolite[1].
나 MOR 제올라이트에서도메탄올에서탄화수소가잘생성되지만,
탄소침적에의한활성저하가빠르다. 세공내에인이나붕소등을 담지하거나산점수를줄여주면올레핀에대한선택성을높이고활
성저하속도를어느정도늦출수있다. Ni, Ca, Pb 이온을일부
교환한 CaY 제올라이트에서는 C2~C4올레핀이많이생성되며, 알
루미늄을제거하여산점수가줄어든 MOR 제올라이트에서는프로
필렌이많이생성된다[5]. 그러나저급올레핀이목적생성물인 MTO
공정에서는큰분자가생성되지않고분자크기가작은저급올레 핀이주로생성되도록 10개산소원자고리로이루어진중간크기
세공(medium pore) 제올라이트와분자체를촉매로사용한다.
MFI 제올라이트에서 MTG 공정의생성물분포는매우특이하여,
탄소개수가 12개이상인탄화수소가생성되지않는다. 이러한생
성물의선택성때문에 MTG 공정의생성물을추가정제없이가솔
린으로쓸수있다. MFI 제올라이트의구부러진세공의길이는탄
소사슬이 12개인탄화수소와길이가거의비슷하다. 긴탄화수소가 구부러진세공을통과하려면심하게구부려져야하므로, C12이상 의탄화수소가생성되기어렵다. 이런이유로 MFI 제올라이트에서 는 C5~C12의가솔린유분만이선택적으로생성된다. 이뿐아니라
C12이상의긴탄화수소, 즉끓는점이높은탄화수소가생성되지않 아서탄소침적에의한활성저하가느려촉매수명이길다. 이와 함께두종류의세공이교차하는공간이제법커서방향족화합물 이나가지달린탄화수소가잘생성되므로 MFI 제올라이트촉매에
서제조한가솔린은옥탄가가높다[1, 6].
MFI 제올라이트에서는파라핀과방향족화합물생성단계까지 반응이진행되므로, MFI 제올라이트를 MTO 공정에촉매로사용하 려면전환반응이저급올레핀생성단계에서중단되도록촉매성
질을조정해야한다[31-34]. Si/Al 몰비를높여산점수를줄이거나,
인이나붕소를담지하여강한산점을중화시킨다. 철이나붕소를
MFI 제올라이트의골격에치환하여도 MTO 반응에서저급올레핀 에대한선택성이높아진다. 세공입구주변에실리카를담지하여 세공입구를줄여도올레핀에대한선택성이향상된다. 세공입구 가줄어들면가지달린탄화수소보다확산이빠른저급올레핀이많 이생성되기때문이다. 다양한방법으로올레핀에대한선택성을높
일수있는점도촉매로서 MFI 제올라이트의큰강점이다.
MFI 제올라이트이외에알루미늄과인으로이루어진분자체
(aluminophosphate)도 MTO 공정에촉매로사용한다[4, 5, 35-41].
이총설에인용된참고문헌의절반정도가 SAPO 분자체를촉매로 언급할정도로이에관련된연구결과가많다. 소량의실리콘이골
격에치환되어산성을띠는 SAPO-34 분자체는 UOP/Hydro MTO
공정의촉매로사용될만큼저급올레핀에대한선택성이우수하다[4].
SAPO-34 분자체는차바자이트(CHA)와골격구조가같으나산성
은약하다. SAPO-34 분자체자체로는 MTO 공정에서저급올레핀에
대한선택성이그리높지않으나, 니켈이들어있는 Ni-SAPO-34 촉
매는메탄올전환율이 100%일때에틸렌에대한선택도가 90%일 정도로아주특이하게높다. 저급올레핀전체에대한선택도는
Fe-silicate에서도높지만, 에틸렌에대한선택도는 Ni-SAPO-34 촉 매에서가장높다. 니켈이골격에치환되어강한산점뿐아니라약 한산점도줄어들어방향족화합물생성이억제되는데따른효과로
높은선택성을설명한다. 실리카함량이높은 SAPO-35 분자체도
MTO 반응에선택성이높다[42]. SAPO-18 분자체는 SAPO-34 분 자체와마찬가지로 MTO 반응에활성과선택성이높다. 또한, 촉매 수명이길고합성에사용하는주형물질이저렴하여촉매비용이절 감된다[43-45]. MTO 공정의촉매에대한설명은 Stöcker의총설에 잘정리되어있으므로이를참고하기바란다[5].
3. 메탄올이 탄화수소로 전환되는 반응의 기구 메탄올이고체산촉매에서디메틸에테르를거쳐탄화수소로전환 되는반응은앞에서언급한대로진행경로는간단하나세부적인 반응기구에대해서는논란이많았다. MTG 공정이처음발표되었
을때는명확한중간체를제시하고이를근거로 MTG 반응의진행
과정을설명하려는시도가대부분이었다. 그러나앞에서이야기한 대로최근에는세공내에생성된탄화수소중간체를활성물질로 보는주장이힘을얻고있다. 아직완벽하지는않지만탄화수소뭉 치의구조와기능이상당부분밝혀졌다는점에서이총설에서는
‘탄화수소활성체반응기구’ 메탄올전환반응을설명한다[9, 14].
메탄올에서탄화수소가생성되는반응의첫단계는메탄올에서 디메틸에테르의생성반응이다. 브뢴스테드산점이메탄올에양성 자를주면양이온중간체가생성되고, 이어아래반응식에쓴대로
메탄올과협동(concerted) 반응을통해디메틸에테르가생성된다고
본다[1].
CH3OH + H+ → CH3OH2+
CH3OH2+ + CH3OH → (CH3)2O + H2O + H+
양이온중간체에서물이떨어져메틸카르베니움이온이생성된후 메탄올의산소에결합하여디메틸에테르가생성된다고볼수도있 다. 그러나메틸카르베니움이온은매우불안정하여생성되기어 려우므로탈수와생성단계가동시에진행되는협동반응이더합 리적이다.
Fig. 3. Variation of product composition with temperature over MFI zeolite[1].
디메틸에테르의생성을메톡시기와관련지어설명하는문헌도
있다[11, 46, 47]. 메탄올이제올라이트표면의브뢴스테드산점에
흡착하여물이제거되면서표면에결합된메톡시기가생성된다. 이 메톡시기는다른메탄올분자와반응하여디메틸에테르를생성한 다. Z는제올라이트골격을나타내며, 두반응이연계되어흡착과
CH3OH + ZOH → (CH3δ+… OH2δ+… ZOδ−)≠ → ZOCH3 +H2O
ZOCH3+ CH3OH → (CH3δ+… CH3OH2δ+… ZOδ−)≠ → ZOH+CH3OCH3
동시에반응이진행될수도있다. 제올라이트에메탄올이흡착되 면메톡시기가생성되는건확실하지만메탄올이디메틸에테르로 바로전환되는높은온도에서는메톡시기를관찰할수없다는논문
도있다[48]. 메탄올과디메틸에테르간평형에대해서도이견이있다.
디메틸에테르의생성단계에대한논란에비하면메탄올전환반 응의핵심단계인디메틸에테르에서탄소-탄소결합이생성되는단
계에대한논란은주장이많아서훨씬복잡하다. Fig. 4에는직접
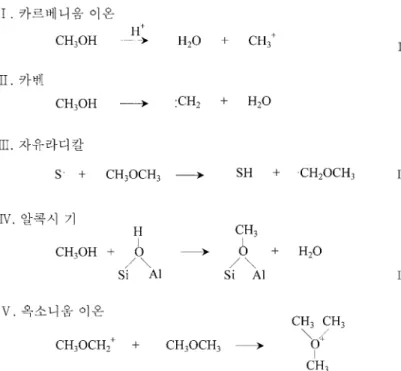
반응기구에서거론하는여러중간체의생성과정을정리하였다[9].
카르베니움이온과카벤이생성되기만하면이들로부터탄소-탄소 결합의생성은용이하여저급올레핀이생성되는데전혀문제가없 다. 그러나메탄올전환반응이진행되는 500oC 이하에서이들처럼 불안정한중간체가서로결합하여에틸렌을만들수있을만큼제올 라이트세공내에많이생성될수있느냐는의문이다. 더욱이물이 같이있는조건에서이런중간체가생성되기가거의불가능하리라는 게일반적인의견이다. 자유라디칼도메탄올전환반응의중간체가 될수있다. 이들이서로결합하여탄소-탄소결합을생성한다고볼 수있으나, 라디칼생성에필요한활성화에너지와이들의추가반 응을위한세공내농도를고려하면가능성이낮다. 메탄올이제올 라이트표면에흡착되면메톡시기등알콕시기가생성된다는점은
잘알려져있다. 그러나메톡시기가메탄올과반응하여에톡시기 가되는알콕시기의사슬성장은아직관찰된예가없어알콕시기 를중간체로보기어렵다. 그러나최근에이르러메톡시기의관여
를주장하는학자도있다. Hunger 등[47]은표면에생성된메톡시기
가디메틸에테르뿐아니라탄소-탄소결합을생성하는활성종이라고 주장하여탄화수소활성체반응기구와다른견해를보인다.
디메틸에테르가제올라이트의산점과반응하여제올라이트골격
에배위한옥소니움이온을만들고, 이들이옥소니움일리드(ylid)로
재배열되면서탄소-탄소결합을형성한다는설명은에너지측면에 서설득력이높다. 직접반응기구에서는옥소니움이온이가장그 럴듯한중간체여서옥소니움이온에서탄화수소가생성되는과정은
세밀히검토되어있다[1]. 이외에도초강산에서생성된카르보니움
이온을중간체로제안한논문도있다. Fig. 4에는중간체의특징만
간략히나타내었으나, 저자에따라중간체의구체적인모양이서로 다르기도한다.
구체적인중간체에근거한반응기구는반응의진행경로와가능 성을검토하는데유리하지만, 실제반응에서관찰되는현상을설명 하는데는적절하지못하다. 직접반응기구의가장큰약점은메탄 올전환반응에서나타나는유도기간을설명하지못하는데있다. 제 올라이트세공내에서중간체가생성되고이들이탄소-탄소결합을 이루면서탄화수소가생성된다면, 중간체의생성과탄화수소로전 환은바로연계되어진행되므로유도기간이나타나지않아야한다.
중간체의종류에따라차이가있으나중간체의세공내에서생성 가능성, 중간체가탄화수소로전환되는반응에서활성화에너지의 크기, 촉매에따른생성물차이, 소량의불순물에의해속도나생성 물분포가크게달라지는현상등을설명하기어려워구체적인중 간체를거치는직접반응기구의설득력에는한계가있다.
이런문제점을극복하기위하여세공내에생성된탄화수소중간 체에근거한반응기구가있다. Fig. 5에는구조는명확하지않지만 세공내생성된탄화수소활성체에서올레핀이생성된다는반응기
Fig. 4. Various intermediates supposed for the methanol conversion
[9]. Fig. 5. Several reaction mechanisms for methanol conversion through
indistinctive intermediates[9].
구를정리하였다. 첫번째반응기구에서는메틸벤젠을중간체로제 안한다. 메탄올이제올라이트세공내에서서로반응하여메틸기가 여러개치환된메틸벤젠이생성된다. 벤젠고리에치환된메틸기 가메탄올에의해알킬화되어에틸기가되고, 이들이제거되면서 에틸렌이생성된다고설명한다. 두번째반응기구에서는중간체로 시클로헥센을설정하였다. 반응진행과정에대한설명은비슷하다.
세번째반응기구는중간체가탄화수소인점은확실하나구조를정 확히알수없다는뜻에서 (CH2)n으로중간체를나타내었다. 지방족 과방향족탄화수소가모두포함된다. 세공내에탄화수소중간체 가형성된후이들로부터탄화수소가생성된다는뜻이다. 구체적인 중간체는제시하지못하여추가반응의진행경로가애매하다는한 계는있으나, 이러한중간체를가정하면메탄올전환반응의유도기 간이나반응조건에따라초기올레핀이달라지는현상, 불순물이 속도에미치는영향등을쉽게설명할수있다. 반응조건에따라 중간체의구조가달라지고, 메탄올에들어있는올레핀이나방향족 불순물이중간체생성을촉진하기때문에반응속도가빨라지고, 또 세공의모양에따라생성되는탄화수소중간체의구조가달라제올 라이트나분자체의종류에따른활성과선택성이다르다고설명할 수있다. 이처럼메탄올전환반응에서나타나는여러현상을설명 하는데는중간체생성을가정하는게아주유용하다. 그러나처음
Haag 등여러연구자들이개별적으로중간체를거치는반응기구를
제안하였을때는중간체의구조를구체적으로제시하지못하였기 때문에중간체구조가확실한직접반응기구에비해중요하게다루 어지지않았다. 그러나반응에서나타나는현상을설명하는데적절 하여배제할수는없었다. 이반응기구에서주장하는 (CHx)n이라고 나타낸중간체에는탄소에비해수소가적다고보아이반응기구를
‘carbon-pool mechanism’이라고불렀다. 그러나이후중간체의구조 가알려지면서앞에서이야기한대로 ‘탄화수소활성체반응기구’
로명칭이달라졌다.
그후중간체의구조에대한연구가 Kolboe 연구팀과 Haw 연구
팀에의해수행되었다[12-21]. 13C으로표시된메탄올이나올레핀과 반응을통해중간체의반응성에대한연구결과도축적되었다. GC-MS
반응기를이용한생성물내동위원소연구는반응기구유추에매우 효과적이었다[49, 50]. 앞서제안된탄화수소활성체반응기구로반 응경향을동위원소분포로설명할수있어, 탄화수소중간체의존 재를간접적으로검증할수있었다. 나아가촉매에공급한메탄올 이반응시간이경과함에따라어떻게변하며, 반응도중촉매내에 는어떠한탄화수소가생성되는지동시조작되는 CP/MAS Solid-
state NMR로직접관찰하였다. 반응생성물로부터메탄올전환반
응을정량적으로해석하면서동시에제올라이트세공에형성되는 물질을확인하므로반응기구를구체적으로유추할수있었다.
NMR 조사결과의예를든다. 673 K에서13C-메탄올 20µl를펄
스로 0.3 g SAPO-34 촉매에가한후시간별로그린 NMR 스펙트
럼을 Fig. 6에보였다[15]. 메탄올을가한후급격히냉각시켜 SAPO-34
분자체에들어있는탄화수소의 NMR 스펙트럼을그렸다. 2초후에 는반응이전혀진행되지않아메탄올의탄소피크만나타난다.
4초후에는골격에결합되어있는메톡시기의탄소피크가 56 ppm
에서나타난다. 이시간대에서는올레핀이아주조금밖에생성되지 않아서, 전환반응의생성물이나타나지않는유도기간이관찰된다.
시간이조금더경과하면 130 ppm 근처에서방향족탄소의피크가,
20 ppm에서는메틸기의탄소피크가관찰되어제올라이트내의메
틸벤젠이생성되었음을보여준다. 시간이더경과하면메틸기의탄소
피크는작아지나방향족탄소의피크는그대로있다. SAPO-34 분자
체의세공은입구가작아서벤젠화합물이통과할수없으므로, 메틸 벤젠은세공내에잡혀있다. 그래서방향족탄소의피크크기는변하 지않는다. 그러나올레핀이생성되면서메틸기탄소의피크는줄어 들어메틸벤젠탄화수소활성체에서올레핀이생성됨을보여준다.
메탄올의첫번째펄스에서는시간이상당히지나야올레핀이생 성되나, 일단메틸벤젠이생성된후에는메탄올펄스를가하면올 레핀이바로생성되어유도기간이없어진다. 세공내에서메탄올전 환반응의활성점으로작용하는메틸벤젠의생성에소요되는시간 이바로유도기간인셈이다. 메틸벤젠의촉매활성점으로서역할을 확인하기위해제올라이트에메틸벤젠을생성시킨후이들의메틸 기치환개수와생성물의연관성을조사하였다[18]. 메틸기가다섯 개치환되어있는펜타메틸벤젠이세공에들어있는촉매에메탄올 을가하면에틸렌이생성된다. 이에반해메틸기가여섯개치환되 어있는헥사메틸벤젠이들어있는촉매에메탄올을가하면프로필 렌이주로생성된다. 트리메틸벤젠에서는에틸렌이주로생성되지 만헥사메틸벤젠에서는프로필렌이훨씬많이생성되어, 세공내에 존재하는메틸벤젠의구조에따라생성물의선택성이결정되었다.
Haw 연구팀에서는탄화수소활성체로메틸기가여러개치환된 폴리메틸벤젠을들지만, 다른연구자들은 C6~C12올레핀과방향족
화합물이섞여있는상태로설명한다[47]. 548~673 K SAPO-34 분
차체촉매에서메탄올전환반응이진행될때그린13C CF MAS NMR
스페트럼에서는폴리메틸벤젠외에도헥센, 옥텐, 헥사디엔등올레 핀과시클로올레핀의피크도나타나기때문이다. 이뿐아니라p-자 일렌등도존재한다고본다. 이들중방향족화합물이메탄올에의 해알킬화되어펜타메틸벤젠이나헥사메틸벤젠이생성되고, 반응조 건에서이들이불안정하여에틸렌이나프로필렌으로분리되면서올 레핀이생성된다고본다. 세공내의올레핀중메틸벤젠만올레핀 생성에관여하는지아니면다른구조의올레핀에서도올레핀이생
Fig. 6.13C NMR spectra of hydrocarbon formed in SAPO-34 molec- ular sieve at 673 K by adding a pulse of 13C-methanol[15].
성되는지여부는확실치않으나주로폴리메틸벤젠을거쳐올레핀 이생성되는점은확실하다.
탄화수소활성체반응기구에서는메틸벤젠탄화수소활성체에서 올레핀이생성되는과정을두가지반응기구로설명한다. 벤젠고 리에치환된메틸기가알킬화되어에틸기가된후이들이떨어지 면서에틸렌이생성되는곁가지알킬화(side chain alkylation) 과정 과고리의수축과팽창단계를거치면서올레핀이생성되는짜내기
(paring) 과정이다. 고리를줄여주면서올레핀을생성한다는뜻으로
‘짜내기반응경로’라고이름붙였다. Fig. 7에대표적인반응과정
을보였다[9].
곁가지알킬화기구에서는메탄올이첨가되면서치환기가길어진
다[18-21]. 헵타메틸벤제니움이온을거쳐치환기의탄소수가늘
어난다고설명하고있으나, 이그림에서는간략하게나타내기위해 이를생략했다. 제올라이트에붙어있는헵타메틸벤젠니움이온을 거친다고해서 ‘exocyclic methylation 경로’라고부르기로한다[14].
벤젠고리에치환되어있는메틸기가많을수록프로필렌이생성될 확률이높아진다.
이에비해짜내기반응에서는벤젠고리가 5각형고리로수축되 면서치환된알킬기의탄소사슬이길어진다. 메탄올이메틸벤젠과 반응하여치환된메틸기수가많아지면벤젠고리가오각형고리로 줄어들고치환된곁가지는에틸기나프로필기로사슬이길어진다.
그래서짜내기반응경로를거치면프로필렌이나부텐이주로생성 되어야한다. 이들이고리에서떨어져서올레핀이생성된후, 메탄올 이다시가해지면고리에메틸기로치환된다. 치환된메틸기가 많아지면고리가팽창되면서벤젠고리가복원된다.
곁가지알킬화반응기구의타당성을검증하기위해올레핀의생 성단계의에너지를이론적인방법으로계산하였다[51]. 메틸벤젠에 서올레핀이생성되리라예상되는경로에서나타날수있는중간체 의엔탈피값을계산하였다. 예상한중간체들의엔탈피값이과도 하게크지않아서이들이생성되는데문제가없다고판단하였다. 또 반응의진행과정에서포텐셜에너지가특히낮은단계가없어진 행경로에도무리가없었다. 계산결과로보면올레핀생성에서곁 가지알킬화기구가짜내기기구보다가능성이크다. 치환된메틸 기의개수가많을수록탄화수소활성체의촉매활성이크고프로필 렌에대한선택성도높다는결과로실험결과와잘일치하였다.
메탄올에서올레핀이생성되는반응의촉매활성점이메틸벤젠이 라는점을검증하기위해메틸벤젠에하이드록실기나불소기가치
환된물질을제올라이트세공내에형성시켰다[9, 52]. 세공내에생 성된메틸벤젠을산소로처리하면페놀유도체가되고, 메탄올을추 가로가하여펜타메틸페놀을만들수있다. 펜타메틸페놀은헥사메 틸벤젠과치환기하나가다르지만, 하이드록실기가치환되어있는 펜타메틸페놀에서는메탄올전환반응이진행되지않는다. 하이드록 실기대신불소기를치환한할로벤젠역시촉매활성점으로작용 하지못한다. 이러한차이는치환된하이드록실기나불소기에의 해곁가지알킬화반응의중간체인헵타메틸벤젠니움이온이생성되 지못하여곁가지알킬화반응이진행되지못하기때문으로설명한다.
이외에도메틸벤젠탄화수소활성체에의해메탄올전환반응이 진행된다는주장을뒷받침하는여러가지실험증거가있다[9, 53].
메탄올에들어있는불순물의영향, 제올라이트세공구조의영향,
에틸렌과프로필렌의초기동시생성물등에서탄화수소활성체의 생성을유추할수있다. 메탄올전환반응에서유도기간은메탄올 의순도에따라크게다르다. 올레핀이나방향족화합물이아주소 량만들어있어도유도기간이아주짧아진다. 이러한물질들은제 올라이트의세공내에서메틸벤젠계탄화수소활성체를빠르게만 들수있다. 아주순수한메탄올을반응물로사용하면유도기간이 아주길어진다. 헥사메틸벤젠이생성되는데시간이많이걸리기때 문이다. 제올라이트세공내에생성되는탄화수소활성체의구조가 생성물에대한선택성을결정한다는사실역시탄화수소활성체반
응기구를지지한다. FER 제올라이트에서는메탄올전환반응의주
생성물이에틸렌과프로필렌이아니고부텐이다. 10개산소원자고 리세공과 8개산소원자고리세공이서로교차하므로교차부분의 공간이좁아메틸벤젠이생성되기어렵기때문으로설명한다. SAPO-34
촉매에서초기생성물로에틸렌과프로필렌이같이생성되는점도 탄화수소활성체반응기구로설명할수있다. 카벤이나카르보니움 이온등구조가확실한중간체에서는초기올레핀으로에틸렌이생 성될수는있으나프로필렌이생성되기는어렵다. 그러나메틸벤젠 이활성체이면에틸렌과프로필렌이같이생성되는현상을메틸벤젠 에 ‘치환된메틸기의개수가달라서’라고설명할수있다.
탄화수소활성체반응기구에서는메탄올전환반응이진행되면 세공내에형성되는탄화수소의종류가달라져서촉매의활성이저
하되는걸로설명한다[53]. 메틸벤젠, 특히헥사메틸벤젠이세공내
에형성되어있으면메탄올전환반응이활발하게진행된다. 그러 나메틸벤젠이산점에서메틸나프탈렌으로골격이성질화되면활성
이낮아진다[17]. 나프탈렌은방향족고리가벤젠고리에비해안정
하여메탄올에의한곁가지알킬화반응이나고리의수축과팽창을 통한짜내기반응이잘진행되지않는다. 메틸나프탈렌에서더안 정한페난트렌이나피렌이생성되면활성체가더욱안정해지고이 들로세공이가득차면반응물과생성물의이동이억제되어촉매로 서기능을상실한다. 세공내에서메틸벤젠→메틸나프탈렌→페난트 렌, 피렌으로반응이진행되어, 이들이점유한세공이많아지면메 탄올전환반응에서촉매활성을잃는다.
Haw의최근총설[53]에서는메틸벤젠탄화수소활성체에의한메
탄올에서올레핀생성기구를그림으로나타내었다. 제올라이트나 분자체의세공내에메틸벤젠이생성되는단계, 헥사메틸벤젠에메 탄올이반응하여곁가지알킬화반응이진행되는단계, 이로부터올 레핀이생성되는단계, 다시메탄올이반응하여헥사메틸벤젠이재
생되는단계가설명되어있다. 원래그림이좀복잡하여 Fig. 8에는
이해하기쉽게단계별로나누어그렸다. 이그림에보인대로개시
Fig. 7. Formation of olefins from methylbenzenes[9].
단계에서는메틸벤젠이생성되고, 올레핀은메틸벤젠과메탄올이반 응하여만들어지며, 활성저하는메틸벤젠이나프탈렌이나피렌으 로구조가달라지는현상과연관지을수있다.
탄화수소활성체반응기구는제안단계에서구체적인중간체가 없고진행과정에대한합리적인설명이없어그의의가과소평가 되었다. 무언가생성되어전환반응의활성점으로작용하지만그실 체를이야기하지못하므로설득력이없었다. 그러나탄화수소활성 체가헥사메틸벤젠으로구체화되면서반응기구로서의의가높아졌 다. 메탄올전환반응의개략적인경로나현상을설명하기충분하다.
부텐의골격이성질화반응에서도탄화수소활성체를활성점으로
보는주장이있다[54]. Guisnet 연구팀에서주장한의사단분자반응
기구(pseudomonomolecular mechanism)로활성점은페리어라이트 세공내에형성된탄화수소뭉치에붙어있는카르베니움양이온이
다. Fig. 9에보인대로세공내에포획된벤젠고리에생성된카르
베니움양이온에서부텐의골격이성질화반응이아주효과적으로 진행된다. 반응중간체의에너지값적정성이나골격이성질화반응 에대한선택성이세공내에탄소가침적되면서같이높아지는현 상은탄화수소뭉치가활성점이라는증거가된다. 이논문에서도탄 화수소활성체반응기구에의해진행되는 MTO 반응과골격이성질 화반응의유사성을설명하고있다. 이런점에서산점으로탄화수 소의전환반응을설명하던단계에서탈피해야할시점이되었다는 생각이든다. 제올라이트세공은탄화수소활성체를생성시키는그 릇으로서작용하고, 이세공내에생성된탄화수소활성체가균일 계촉매처럼전환반응을촉진하는지모르기때문이다.
4. MTO 공정에 대한 전망
물가상승에도불구하고원유가격은상당히오랜기간배럴당
30달러이하로유지되어서, 원유외다른원료에서저급올레핀을
생산하려는시도에관심이낮을수밖에없었다. 그러나 2006년들 어서우리나라의주요수입원인두바이유의가격이배럴당 70달러 를넘어서서에너지가사회문제로부각되어에너지절약이나원유
대체자원의확보에관심이높아졌다. MTO 공정의가격경쟁력을
검토하던시점[3]에비해최근원유가격이급등하기는했어도, 그
동안의물가상승폭을감안하여 MTO 공정의경제적타당성에의
문을제기하는전문가도있다. 또대형메탄올공장이가동되고있 어도메탄올의안정적공급에대한우려도무시할수없다. 그러나 원유가격의지속적상승은메탄올에서올레핀을생산하는공정의 필요성을강조하고있다. 원유가격의상승에따른충격을줄이면 서석유화학산업의지속적발전을기하는데도움이될수있기때 문이다.
MTO 공정에대한촉매개발도시도해볼만한과제이다. MTG
공정에서는 MFI 제올라이트가거의독보적이지만, MFI 제올라이
트외에 SAPO계촉매도 MTO 공정에서는우수하다. 인담지나골
격원소치환으로 MFI 제올라이트의저급올레핀에대한선택성을
높일수도있고, 촉매의형태를바꾸어성능을개선할수도있다. 세 라믹폼(ceramic foam)에 MFI 제올라이트를도포하거나[55] 막으
로만든촉매의성능이더우수하다[56]. 제올라이트막에서산점의
분포상태를조절하여도올레핀에대한선택성이높아진다[57].
SAPO-34나 SAPO-18 등분자체도 MTO 공정에촉매로서적절하
며, Co, Mn, Ni 등전이금속을치환하면촉매성질이크게달라진
다. 제올라이트와분자체의종류, 산점의농도와세기, 입자크기,
전이금속등을골격원소의종류등에따라저급올레핀에대한선 택성과촉매수명이크게달라져서촉매개발의가능성이상대적으
로높다. SAPO-34 촉매만해도초기에는저급올레핀에대한선택
성은높으나탄소침적에의한활성저하가심하여촉매로서적용 이우려되었다. 그러나재생공정을도입하고반응물에물을첨가 하거나골격에전이금속을치환하는등으로꾸준한연구성과로 현재는개발되어있는촉매중에서가장우수한촉매로평가되고
있다. SAPO-34를촉매로사용하는 UOP/Hydro MTO 공정에서에
틸렌과프로필렌의수율합은메탄올이거의전환된조건에서 80% 에이르나, 최근기술의발전으로에틸렌과프로필렌의선택도합 이 85~90%에달한다[58]. 프로필렌과에틸렌의생성비도 2.0 이상 까지높일정도로공정제어기술도개선되었다.
Fig. 8. Initiation, propagation, and deactivation of methanol-to-olefins process explained by hydrocarbon pool mechanism[53].
Fig. 9. Reaction mechanism of the skeletal isomerization of n-butene over FER zeolite [54].
반응기구에대한이해가깊어지면더합리적인방법으로 MTO
공정에적절한촉매를설계할수있다. 세공구조의탐색, 골격치 환금속의선정, 반응기나반응조건의선택등이체계적이고합리 적으로진행될수있기때문이다. 표면에형성된알콕시기의반응 참여, 불순물의효과등에대한논쟁이아직도진행되고있어우수
한촉매의개발가능성이아직남아있음을시사한다[59-62]. 동위
원소를활용한반응기구고찰[59]과고체 NMR 기법에동시조작개
념을도입하여세공내물질을분석하므로중간체를규명[11, 63-65]
하여 ‘탄화수소활성체반응기구’가정립되었다. 이제는이를넘어 서서기상반응에서벤젠니움양이온의반응을통해메탄올에서탄 화수소생성과정을조사하거나[66] 메틸할라이드의반응을통해중 간활성체의상태를규명하려는시도가이어지고있다[67]. 디메틸 에테르에서탄소-탄소결합이생성되는단계에대한이론적실험적
연구는계속되고있다[68, 69]. 이러한연구결과를바탕으로반응
기구를더자세히이해하게되면헥사메틸벤젠등탄화수소활성체 가메탄올전환반응의활성점으로효과적으로작용할수있도록 세공구조를조정하고, 활성체의기능을극대화시키는증진제도선 정할수있으며, 세공구조나활성체의손상을최소화하는재생방 법을개발하여촉매의성능을크게증진시킬수있는방안을모색 할수있다. 집중적인연구를통해우리나름의독자적인촉매개발 도기대해본다.
참고문헌
1. Chang, C. D., “Hydrocarbons from Methanol,”Catal. Rev. Sci.
Eng., 25(1), 1-118(1983).
2. Chang, C. D., “Methanol Conversion to Light Olefins,”Catal.
Rev. Sci. Eng., 26(3&4), 323-325(1984).
3. Seo, G., “Conversion of Methanol to Chemical Feedstock and Fuel,”Prog. Ind. Chem., 27(1), 15-22(1987).
4. Keil, F. J., “Methanol-to-Hydrocarbons: Process Technology,” Micropor.
Mesopor. Mater., 29, 49-66(1999).
5. Stöcker, M., “Methanol-to-Hydrocarbons: Catalytic Materials and Their Behavior,”Micropor. Mesopor. Mater., 29, 3-48(1999).
6. Maxwell, I. E. and Stork, W. H. J., Stud. Surf. Sci. Catal., 137
(Introduction to Zeolite Science and Practice, van Bekkum et al.
Eds.), 747-819(2001).
7. Park, J. W., Kim, J.-H. and Seo, G., “The Effect of Pore Shape on the Catalytic Performance of Zeolites in the Liquid-Phase Deg- radation of HDPE,”Pol. Degrad. Stab., 76, 495-501(2002).
8. Liu, Z., Sun, C., Wang, G., Wang, Q. and Cai, G., “New Progress in R&D of Lower Olefin Synthesis,”Fuel Process. Technol., 62, 161-172(2000).
9. Haw, J. F., Song, W., Marcus, D. M. and Nicholas, J. B., “The Mechanism of Methanol to Hydrocarbon Catalysis,”Acc. Chem.
Res., 36, 317-326(2003).
10. Mikkelsen, Ø., Rønning, P. O. and Kolboe, S., “Use of Isotopic Labeling for Mechanistic Studies of the Methanol-to-Hydrocar- bons Reaction. Methylation of Toluene with Methanol over H-ZSM- 5, H-Mordenite and H-Beta,”Micropor. Mesopor. Mater., 40, 95- 113(2000).
11. Hunger, M., “In Situ NMR Spectroscopy in Heterogeneous Cataly- sis,”Catal. Today, 97, 3-12(2004).
12. Dahl, I. M. and Kolboe, S., “On the Reaction Mechanism for
Propene Formation in the MTO Reaction over SAPO-34,”Catal.
Lett., 20(3-4), 329-336(1993).
13. Arstad, B. and Kolboe, S., “The Reactivity of Molecules Trapped within the SAPO-34 Cavities in the Methanol-to-Hydrocarbons Reaction,”J. Am. Chem. Soc., 123, 8137-8138(2001).
14. Olsbye, U., Bjørgen, M., Svelle, S., Lillerud, K.-P. and Kolboe, S., “Mechanistic Insight into the Methanol-to-Hydrocarbons Reac- tion,”Catal. Today,106, 108-111(2005).
15. Song, W., Haw, J. F., Nicholas, J. B. and Heneghan, C. S., “Meth- ylbenzenes are the Organic Reaction Centers for Methanol-to-Ole- fin Catalysis on HSAPO-34,”J. Am. Chem. Soc., 122, 10726-10727 (2000).
16. Haw, J. F., Nicholas, J. B., Song, W., Deng, F., Wang, Z., Xu, T.
and Heneghan, C. S., “Roles for Cyclopentenyl Cations in the Synthesis of Hydrocarbons from Methanol on Zeolite Catalyst HZSM-5,”J. Am. Chem. Soc., 122, 4763-4775(2000).
17. Song, W., Fu, H. and Haw, J. F., “Selective Synthesis of Meth- ylnaphthalenes in HSAPO-34 Cages and their Function as Reac- tion Centers in Methanol-to-Olefin Catalysis,”J. Phys. Chem. B,
105, 12839-12843(2001).
18. Song, W., Fu, H. and Haw, J. F., “Supramolecular Origins of Product Selectivity for Methanol-to-Olefin Catalysis on HSAPO- 34,”J. Am. Chem. Soc., 123, 4749-4754(2001).
19. Song, W., Nicholas, J. B. and Haw, J. F., “A Persistent Carbe- nium Ion on the Methanol-to-Olefin Catalyst HSAPO-34: Ace- tone Shows the Way,”J. Phys. Chem. B, 105, 4317-4323(2001).
20. Sassi, A., Wildman, M. A. and Haw, J. F., “Reactions of Butyl- benzene Isomers on Zeolite HBeta: Methanol-to-Olefins Hydro- carbon Pool Chemistry and Secondary Reactions of Olefins,”J.
Phys. Chem. B, 106, 8768-8773(2002).
21. Sassi, A., Wildman, M. A., Ahn, H. J., Prasad, P., Nicholas, J. B.
and Haw, J. F., “Methylbenzene Chemistry on Zeolite HBeta: Multi- ple Insights into Methanol-to-Olefin Catalysis,”J. Phys. Chem. B,
106, 2294-2303(2002).
22. Park, T.-Y. and Froment, G. F., “Kinetic Modeling of the Meth- anol to Olefins Process. 2. Experimental Results, Model Dis- crimination, and Parameter Estimation,”Ind. Eng. Chem. Res.,
40, 4187-4196(2001).
23. Alwahabi, S. M. and Froment, G. F., “Conceptual Reactor Design for the Methanol-to-Olefins Process on SAPO-34,”Ind. Eng. Chem.
Res.,43,5112-5122(2004).
24. Wu, X., Abraha, M. G. and Anthony, R. G., “Methanol Conver- sion on SAPO-34: Reaction Condition for Fixed-Bed Reactor,”
Appl. Catal. A: General, 260,63-69(2004).
25. Gayubo, A. G., Vivanco, R., Alonso, A., Valle, B. and Aguayo, A. T., “Kinetic Behavior of the SAPO-18 Catalyst in the Trans- formation of Methanol into Olefins,”Ind. Eng. Chem. Res., 44, 6605-6614(2005).
26. Keil, F. J., Hinderer, J. and Garayhi, A. R., “Diffusion and Reac- tion in ZSM-5 and Composite Catalysts for the Methanol-to-Ole- fins Process,”Catal. Today, 50, 637-650(1999).
27. Chen, D., Rebo, H. P. and Holmen, A., “Diffusion and Deactiva- tion During Methanol Conversion over SAPO-34: A Percolation Approach,”Chem. Eng. Sci., S4, 3465-3473(1999).
28. Chen, D., Rebo, H. P., Gronvold, A., Moljord, K. and Holmen, A., “Methanol Conversion to Light Olefins over SAPO-34: Kinetic Modeling of Coke Formation,”Micropor. Mesopor. Mater., 35-
36, 121-135(2000).
29. Dewaele, O., Geers, V. L., Froment, G. F. and Marin, G. B., “The Conversion of Methanol to Olefins: A Transient Kinetic Study,”
Chem. Eng. Sci., 54, 4385-4395(1999).
30. Soundararajan, S., Dalai, A. K. and Berruti, F., “Modeling of Methanol to Olefins (MTO) Process in a Circulating Fluidized Bed Reactor,”Fuel, 80, 1187-1197(2001).
31. Seo, G., Song, Y. S., Byun, D. H. and Ha, B. H., “Olefin Selec- tivity in the Conversion of Methanol over Phosphorus Modified HZSM-5 Catalysts,”Korean Chem. Eng. Res., 26(6), 591-589(1988).
32. Seo, G. and Ryoo, R., “31P, 27Al, and 129Xe NMR Study of Phos- phorus-Impregnated HZSM-5 Zeolite Catalysts,”J. Catal., 124, 224-230(1990).
33. Seo, G., Kim, D. C., Ko, T.-S. and Park, T. J., “The Effect of Phosphorus Impregnation on HZSM-5 Zeolite in 1-Butenes Con- version,”Korean Chem. Eng. Res., 29(4), 494-502(1991).
34. Rhee, K., Cho, M., Jeong, B. and Seo, G., “Catalytic Properties of Borosilicate in Methanol Conversion,”J. Korean Chem. Soc.,
34(4), 360-369(1990).
35. Djieugoue, M.-A., Prakash, A. M. and Kevan, L., “Catalytic Study of Methanol-to-Olefins Conversion in Four Small-Pore Silicoalmino- phosphate Molecular Sieves: Influence of the Structural Type, Nickel Incorporation, Nickel Location, and Nickel Concentration,”J. Phys.
Chem. B, 104, 6452-6461(2000).
36. Chen, D., Moljord, K., Fuglerud, T. and Holmen, A., “The Effect of Crystal Size of SAPO-34 on the Selectivity and Deactivation of the MTO Reaction,”Micropor. Mesopor. Mater., 29, 191-203 (1999).
37. Wilson, S. and Barger, P., “The Characteristics of SAPO-34 which Influence the Conversion of Methanol to Light Olefins,” Micropor.
Mesopor. Mater., 29, 117-126(1999).
38. Dahl, I. M., Modtad, H., Akporiaye, D. and Wendelbo, R., “Struc- tural and Chemical Influences on the MTO Reaction: A Compar- ison of Chabazite and SAPO-34 as MTO Catalysts,” Micropor.
Mesopor. Mater., 29, 185-190(1999).
39. Ko, T.-S. and Seo, G., “Methanol Conversion over SAPO-34 Molecular Sieve Catalyst,”Korean Chem. Eng. Res., 28(2) 163- 171(1990).
40. Ko, T.-S. and Seo, G., “Conversion of Olefins over SAPO-34 Molecular Sieve Catalyst,”Korean Chem. Eng. Res., 29(3), 263- 269(1991).
41. Dubois, D. R., Obrzut, D. L., Liu, J., Thundimadathil, J., Adek- kanattu, P. M., Guin, J. A., Punnoose, A. and Seehra, M. S.,
“Conversion of Methanol to Olefin over Cobalt-, Manganese- and Nickel-Incorporated SAPO-34 Molecular Sieves,”Fuel Pro- cess. Technol., 83, 203-218(2003).
42. Venkatathri, N., “Synthesis and Characterization of High Silica Content Silicoaluminophosphate SAPO-35 from Non-Aqueous Medium,”Catal. Commun., 7, 773-777(2006).
43. Franklin, I. L., Beale, A. M. and Sankar, G., “On the Activity, Longevity and Recyclability of Mn(II) and Co(II) Substituted AlPO-18 Catalysts for the Conversion of Methanol to Light Ole- fins,”Catal. Today, 81, 623-629(2003).
44. Gayubo, A. G., Aguayo, A. T., Alonso, A., Atutxa, A. and Bilbao, J., “Reaction Scheme and Kinetic Modelling for the MTO Pro- cess over a SAPO-18 Catalyst,”Catal. Today, 106, 112-117(2005).
45. Aguayo, A. T., Gayubo, A. G., Vivanco, R., Alonso, A. and Bil-
bao, J., “Initiation Step and Reactive Intermediates in the Trans- formation of Methanol into Olefins over SAPO-18 Catalyst,”Ind.
Eng. Chem. Res., 44, 7279-7286(2005).
46. Wang, W., Seiler, M. and Hunger, M., “Role of Surface Meth- oxy Species in the Conversion of Methanol to Dimethyl Ether on Acidic Zeolites Investigated by In Situ Stopped-Flow MAS NMR Spectroscopy,”J. Phys. Chem. B, 105, 12553-12558(2001).
47. Wang. W., Jiang, Y. and Hunger, M., “Mechanistic Investigation of the Methanol-to-Olefin(MTO) Process on Acidic Zeolite Cat- alysts by In Situ Solid-State NMR Spectroscopy,”Catal. Today,
113, 101-114(2006).
48. Carlson, L. K., Isbester, P. K. and Munson, E. J., “Study of the Conversion of Methanol to Dimethyl Ether on Zeolite HZSM-5 Using In Situ Flow MAS NMR,”Solid State Nucl. Magn. Reson.,
16, 93-102(2000).
49. Dahl, I. M. and Kolboe, S., “On the Reaction Mechanism for Hydrocarbon Formation from Methanol over SAPO-34,”J. Catal.,
149, 458-464(1994).
50. Dahl, I. M. and Kolboe, S., “On the Reaction Mechanism for Hydrocarbon Formation from Methanol over SAPO-34. 2. Iso- tope Labelling Studies of the Co-Reaction of Propene and Meth- anol,”J. Catal., 161, 304-309(1996).
51. Arstad, B., Nicholas, J. B. and Haw, J. F., “Theoretical Study of the Methylbenzene Side-Chain Hydrocarbon Pool Mechanism in Methanol to Olefin Catalysis,”J. Am. Chem. Soc., 126, 2991-3001 (2004).
52. Marcus, D. M., Song, W., Abubakar, S. M., Jani, E., Sassi, A.
and Haw, J. F., “Reactions of Halobenzenes with Methanol on the Microporous Solid Acids HBeta, HZSM-5, and HSAPO-5:
Halogenation does not Improve the Hydrocarbon Pool,”Lang- muir, 20, 5946-5951(2004).
53. Haw, J. F. and Marcus, D. M., “Well-Defined (Supra)Molecular Structures in Zeolite Methanol-to-Olefin Catalysis,”Top. Catal.,
34(1-4), 41-48(2005).
54. Guisnet, M., “‘Coke’ Molecules Trapped in the Micropores of Zeolites as Active Species in Hydrocarbon Transformations,”J.
Mol. Catal. A: Chem., 182-183, 367-382(2002).
55. Patcas, F. C., “The Methanol-to-Olefins Conversion over Zeo- lite-Coated Ceramic Foams,”J. Catal., 231, 194-200(2005).
56. Masuda, T., Asanuma, T., Shouji, M., Mukai, S. R., Kawase, M.
and Hashimoto, K., “Methanol to Olefins using ZSM-5 Zeolite Catalyst Membrane Reactor,”Chem. Eng. Sci., 58, 649-656(2003).
57. Tago, T., Iwakai, K., Morita, K., Tanaka, K. and Masuda, T.,
“Control of Acid-Site Location of ZSM-5 Zeolite Membrane and its Application to the MTO Reaction,”Catal. Today, 105, 662-666 (2005).
58. Chen, J. Q., Bozzano, A., Glover, B., Fuglerud, T. and Kvisle, S.,
“Recent Advancements in Ethylene and Propylene Production Using the UOP/Hydro MTO Process,”Catal. Today, 106, 103- 107(2005).
59. Bjørgen, M., Olabye, U., Petersen, D. and Kolboe, S., “The Meth- anol-to-Hydrocarbons Reaction: Insight into the Reaction Mech- anism from [12C]Benzene and [13C]Methanol Coreactions over Zeolite H-Beta,”J. Catal., 221, 1-10(2004).
60. Wang, W., Buchhlolz, A., Seiler, M. and Hunger, M., “Evidence for an Initiation of the Methanol-to-Olefin Process by Reactive Surface Methoxy Groups on Acidic Zeolite Catalysts,”J. Am. Chem.
Soc., 125, 15260-15267(2003).
61. Lesthaeghe, D., Speybroeck, V. V., Marin, G. B. and Waroquier, M., “What Role do Oxonium Ions and Oxonium Ylides Play in the ZSM-5 Catalyzed Methanol-to-Olefin Process,”Chem. Phys.
Lett., 417, 309-315(2006).
62. Jiang, Y., Marthala, V. R. R., Huang, J., Sulikowski, B. and Hun- ger, M., “Effect of Organic Impurities on the Hydrocarbon For- mation via the Decomposition of Surface Methoxy Groups on Acidic Zeolite Catalysts,”J. Catal., 238, 21-27(2006).
63. Seiler, M., Schenk, U. and Hunger, M., “Conversion of Metha- nol to Hydrocarbons on Zeolite HZSM-5 Investigated by In Situ MAS NMR Spectroscopy under Flow Conditions and On-Line Gas Chromatography,”Catal. Lett., 62, 139-1451(1999).
64. Hunger, M., Seilver, M. and Buchholz, A., “In Situ MAS NMR Spectroscopic Investigation of the Conversion of Methanol to Olefins on Silicoaluminophosphates SAPO-34 and SAPO-18 under Continuous Flow Conditions,”Catal. Lett., 74(1-2), 61-68(2001).
65. Han, X., Yan, Z., Zhang, W. and Bao, X., “Applications of In Situ NMR in Catalytic Processes of Organic Reactions,”Curr. Org.
Chem., 5, 1017-1037(2001).
66. Svelle, S., Bjorgen, M., Kolboe, S., Kuck, D., Letzel, M., Olsbye, U., Sekiguchi, O. and Uggerud, E., “Intermediates in the Meth- anol-to-Hydrocarbons (MTH) Reaction: a Gas Phase Study of the Unimolecular Reactivity of Multiply Methylated Benzenium Cations,”Catal. Lett., 109(1-2), 25-35(2006).
67. Svelle, S., Aravinthan, S., Bjorgen, M., Lillerud, K.-P., Kolboe, S., Dahl, I. M. and Olsbye, U., “The Methyl Halide to Hydrocar- bon Reation over H-SAPO-34,”J. Catal., 241, 243-254(2006).
68. Lesthaeghe, D., Speybroeck, V. V., Marin, G. B. and Waroquier, M., “Understanding the Failure of Direct C-C Coupling in the Zeolite-Catalyzed Methanol-to-Olefin Process,”Angew. Chem. Int.
Ed., 45, 1714-1719(2006).
69. Marcus, D. M., McLachlan, K. A., Wildman, M. A., Ehresmann, J.
O., Kletnieks, P. W. and Haw, J. F., “Experimental Evidence from H/D Exchange Studies for the Failure of Direct C-C Cou- pling Mechanism in the Methanol-to Olefin Process Catalyzed by HSAPO-34,”Angew. Chem. Int. Ed., 45, 3133-3136(2006).

![Fig. 3. Variation of product composition with temperature over MFI zeolite[1].](https://thumb-ap.123doks.com/thumbv2/123dokinfo/5408315.220524/4.892.67.428.110.511/fig-variation-product-composition-temperature-mfi-zeolite.webp)
![Fig. 6. 13 C NMR spectra of hydrocarbon formed in SAPO-34 molec- molec-ular sieve at 673 K by adding a pulse of 13 C-methanol[15].](https://thumb-ap.123doks.com/thumbv2/123dokinfo/5408315.220524/6.892.517.773.134.466/spectra-hydrocarbon-formed-sapo-molec-molec-adding-methanol.webp)
![Fig. 7. Formation of olefins from methylbenzenes[9].](https://thumb-ap.123doks.com/thumbv2/123dokinfo/5408315.220524/7.892.65.429.146.341/fig-formation-of-olefins-from-methylbenzenes.webp)
![Fig. 9. Reaction mechanism of the skeletal isomerization of n -butene over FER zeolite [54].](https://thumb-ap.123doks.com/thumbv2/123dokinfo/5408315.220524/8.892.65.434.134.485/fig-reaction-mechanism-skeletal-isomerization-butene-fer-zeolite.webp)
