85 책임저자:고경혁, 660-751, 경상남도 진주시 칠암동 92
경상대학교 의과대학 병리학교실 Tel: 055-751-8761, Fax: 055-759-7952 E-mail: [email protected]
접수일:2008년 5월 22일, 게재승인일:2008년 6월 18일
Correspondence to:Gyung Hyuck Ko
Department of Pathology, College of Medicine, Gyeongsang National University, 92, Chilam-dong, Jinju 660-751, Korea
Tel: +82-55-751-8761, Fax: +82-55-759-7952 E-mail: [email protected]
p16
INK4aPromoter Methylation and
Immunohistochemical Expression in Breast Carcinoma and Association with Pathological Features
Jong Sil Lee, Dae Hyun Song, Jung Wook Yang, Dong Chool Kim, Jeong Hee Lee and Gyung Hyuck Ko Department of Pathology, College of Medicine, Gyeongsang National University, Jinju 660-751, Korea A cyclin-dependent kinase (CDK) 4 inhibitor (p16INK4) gene located at 9p21 has been shown to be inactivated in a variety of tumors by homozygous deletions, point mutations, or hypermethylation of its promoter region. The etiologic role of the cdk inhibitor p16INK4a tumor suppressor protein in breast cancer remains unclear. This study attempts to assess the methylation pattern of p16INK4a gene and its correlation with the expression of p16INK4a and to determine, by comparing the correlation between other standard immunohistochemical parameters (Cerb B2, estrogen receptor, progesteron receptor, p53, and Ki-67) and clinicopathological features, the significance of p16INK4a methylation and p16INK4a immunohistochemical expression in breast carcinoma. Immunohistochemical p16INK4a expression was found in 21 (42%) of 50 cases. There was a correlation between loss of p16INK4a expression and advanced stage (p=.016). p16INK4a promoter methylation was found in 72%. Twenty-three (79.3%) of 29 tumors with loss of expression of p16INK4a in immunohistochemistry showed DNA methylation of the p16INK4a gene. Loss of p16INK4a expression was related with high stage breast tumors and p16INK4a promoter hypermethylation was the main mechanism of loss of p16INK4a expression. (Cancer Prev Res 13, 85-90, 2008)
Key Words: p16INK4a, Methylation, Immunohistochemistry, Breast cancer
INTRODUCTION
Breast cancers are generally considered to result from the accumulation of multiple clonal changes in genes that regulate cell growth and differentiation. One of the mechanism involved in the carcinogenesis process is loss of tumor suppressor gene function, which normally acts as a negative regulator of cell proliferation.1) Tumor suppressor genes participate in a variety of critical and highly conserved cell functions, including regulation of the cell cycle and apoptosis, differentiation, sur- veillance of genomic integrity and repair of DNA errors, signal transduction, and cell adhesion.
In vitro, p16INK4a promoter methylation has been shown to result in transcriptional silencing of gene.1) Hypermethylation
of selected CpG site within CpG island in the promoter region of genes is associated with loss of gene expression and is observed in both physiological conditions, such as X chro- mosome inactivation, and pathologic conditions such as neo- plasia. In recent years, it has been increasingly recognized that the promoter regions of some genes, whose activity is governed by CpG islands, may be methylated to varying degrees in multiple types of human cancer.2,3) The etiologic role of the cyclin-dependent kinase (CDK) inhibitor p16INK4a tumor suppressor protein in breast cancer remains unclear. A CDK4 inhibitor (p16INK4a) gene located at 9p21 has been shown to be inactivated in a variety of tumors by homozygous deletions, point mutations, or hypermethylation of its promoter region.
It binds preferentially to CDK4/6 and prevents their association with D-type cyclins, thus inhibiting pRB phosphorylation and
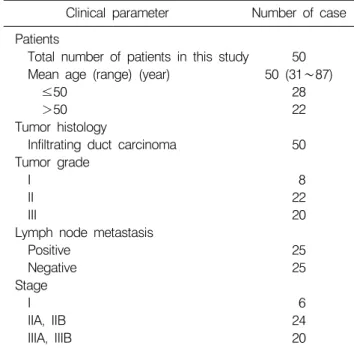
Table 1. Clinicopathological details of cases used in this study Clinical parameter Number of case Patients
Total number of patients in this study 50 Mean age (range) (year) 50 (31∼87)
≤50 28
>50 22
Tumor histology
Infiltrating duct carcinoma 50 Tumor grade
I 8
II 22
III 20
Lymph node metastasis
Positive 25
Negative 25
Stage
I 6
IIA, IIB 24
IIIA, IIIB 20
progression through the cell cycle. Some study showed mutations or deletions of the p16INK4a gene are uncommon in breast cancer,3,4,5) and hypermethylation of the p16INK4a pro- moter region has been described as an alternative mechanism to the observed lack of p16INK4a protein expression in a proportion of breast tumors.6,7)
This study attempts to assess, by Methylation-Specific Polymerase Chain Reaction (MSP), the methylation pattern of p16INK4a gene and its correlation with the expression of p16INK4a and to determine, by comparing the correlation between other standard immunohistochemical parameters (Cerb B2, ER, PR, p53, and Ki-67) and clinicopathological features, the signi- ficance of p16INK4a methylation and p16INK4a immunohisto- chemical expression in breast carcinomas.
MATERIALS AND METHODS 1. Specimens
Fifty specimens of breast carcinoma were collected from the Gyeongsang National University hospital, from 2004 to 2007.
The patients who had undergone a modified radical mastec- tomy or a wide excision at the surgery department and who had been diagnosed as having infiltrative duct carcinoma at the pathology department were included in this study. None of the patients had received radiotherapy or chemotherapy before surgery. All the patients were advised of procedures and provided written informed consent. The histopathological diagnoses of the tumors were described according to the WHO International Classification of Disease for Oncology.8) The clinical staging was determined by the TNM Staging System.
The malignancy of infiltrating duct carcinomas was scored according to the Scarff-Bloom-Richardson classification.9) The ages of the patients ranged from 31 to 87 (mean 50) years old. Details are presented in Table 1.
2. Immunohistochemistry
Standard immunohistochemical detection with minor modi- fications, was performed on paraffin-embedded tissue sections fixed with buffered formalin. In this study, we optimized our staining protocol by using antibody dilution giving the best ratio between low background and high specific staining.
Sections of 4μm thickness were mounted on poly-L-lysine coated slides, deparaffinized and hydrated through graded alcohols to water. The slides were microwaved in citrate buffer
for 10 min in an 800 W microwave oven for antigen retrieval, then allowed to cool for 20 min. Protein expression was studied using specific antibodies against the estrogen receptor (ER) (monoclonal mouse anti-human ER 1D5, Dako, Carpinteria, CA, USA, 1:20 dilution), the progesterone receptor (PR) (monoclonal mouse anti-human progesterone receptor 1A6, Dako, Carpinteria, CA, USA, 1:20 dilution), p53 (monoclonal mouse anti-human p53 protein clone DO-7, Dako, dilution 1:2,000), Ki-67 (monoclonal mouse anti-human ki-67 anti- gen clone MIB-1, Dako, dilution 1:2,000), Cerb B2 (rabbit monoclonal anti-human cerb B2 oncoprotein, Neomarkers, dilution 1:20) and p16INK4a (Dako, Carpinteria, CA, USA, dilution 1:400). Tumors were classified by intensity of stain- ing and the percentage of cells showing antibody reactivity.
The ER, progesteron receptor (PR), and p53 sections were scored for the immunohistochemical signal as follows: strong (2+) when more than 10% of tumor cells are stained, weak (1+) when not more than 10% of tumor cells were stained, and absent (0) when no tumor cells were stained. A tumor was considered positive for Ki-67 when more than 25% of tumor cells were immuno- histochemically stained for Ki-67. For Cerb B2, the strength of the membranous staining was recorded by a four-step scale 0, 1+ to 3+ as follows: strong (3+) when the membranes of all tumor cells were stained continuously, weak (2+) when the membranes of not all but more than 10%
of tumor cells were stained continuously, focal (1+) when the
membranes of more than 10% of tumor cells were stained discontinuously, negative (0) when less than 10% of infiltrating tumor cells were stained. Estimating 10 high power fields, nuclear and cytoplasmic staining was identified as positive staining for p16. The intensity of staining was scored on a scale of 0 to 2+. A tumor was considered negative for p16 when not more than 5% of tumor cells were weakly (<1+) stained for p16.
3. DNA extraction
Genomic DNA was extracted from formalin-fixed, paraffin- embedded samples using EX-WAX DNA extraction kit (Che- micon), according to the manufacturer's instructions. This kit was intended to extract DNA from paraffin-embedded tissue fixed in 10% formalin. DNA was solubilized, while digested proteins were "salted out" and spun to the bottom of the tube.
DNA was precipitated, dried under vacuum, and resuspended.
Once in solution, the DNA was suitable for amplification by PCR. The concentration of DNA was measured in a Bio- Photometer (Eppendorf, Hamburg, Germany) at a wavelength of 260 nm.
4. p16INK4a promoter methylation and Methyla- tion-specific PCR
The methylation status of the p16INK4a promotor was determined by methylation-specific PCR (MSP). MSP can rapidly assess the methylation status of virtually any group of CpG sites within a CpG island, independent of the use of cloning or methylation-sensitive restriction enzymes. The assay consists of initial modification of DNA by sodium bisulfite, converting all unmethylated, but not methylated, cytosines to uracil, and subsequent amplification with primers specific for methylated versus unmethylated DNA. MSP requires very small quantities of DNA, is sensitive to 0.1% methylated alleles of a given CpG island locus, and can be performed in DNA extracted formalin-fixed, paraffin-embedded samples.
Bisulfite processing of DNA was performed in principle as described by Frommer et al.10) and the modifications introduced by Clark et al.11) The p16INK4a methylation was examined using a CpGenomeTM p16 Modification Kit (Q Biogene Heidelberg, Germany) according to manufacturer's instruction. In brief, MSP requires an initial bisulfite reaction in which all unme- thylated cytosines are deaminated and converted to uracils, while 5-methylcytosines remain unaltered. Modified DNA was
used as a template for MSP using primers specific for either methylated or modified unmethylated sequences. Primer se- quences were as follows: p16INK4a Unmethylated (F) 5'-TTA- TTAGAGGGTGGGGTGGATTGT-3' and p16INK4a Unmethy- lated (R) 5'-CAACCCCAAACCCACAACCATAA-3', p16INK4a Methylated (F) 5'-TTATTAGAGGGTGGGGCGGATCGC-3' and p16INK4a Methylated (R) 5'-GACCCCCGAACCGCGA- CCCTAA-3'. A set reaction conditions involved treatment of the DNA with 3.1M sodium bisulfite (pH 5.0) at 55o for 16 h. The parameter of PCR cycle was: 95o 10 min, 95oC 45 s, 55oC 45 s, 72oc 60s, 72oC 3 min. These primers were designed to target the regions of the p16INK4a promoter which initially undergo methylation in model systems. The annealing tempe- rature for both methylated and unmethylated reactions was 55oC. All of the MSP reactions used 1.5 mM MgCl2, 200 M dNTPs, 10 pmol of each primer, GeneAmp PCR buffer, and 2.5 units of Hot start enzyme (Roche FastStart Taq). Finally, 2μl of DNA was added to a total volume of 50μl. Hot start enzyme was used for PCR to discriminate unmethylated/
methylated DNA. A product of 154 bp produced after PCR using a U primer set indicates that the sample DNA was originally unmethylated, while a product of 145 bp produced using an M primer set indicates that the DNA was methylated.
Positive controls and negative water controls were included for both unmethylated and methylated reactions. PCR products were resolved on 2% agarose gels and stained with ethidium bromide.
5. Statistical analysis
Statistical analysis of correlation of p16INK4a methylation with immunohistochemical expressions and clinicopathological features was preformed with Fisher exact test (2-sided) using SPSS v.12.0 (SPSS, Chicago, III). p values of less than .05 were considered significant.
RESULTS
Immunohistochemical p16INK4a expression was found in 21 (42%) of 50 cases (Fig. 1A). There were loss of p16INK4a protein expression in 58% (Fig. 1B). When the p16INK4a expression was compared with clinicopathological variables, there was a cor- relation between loss of p16INK4a expression and advanced stage (p=.016). There was no correlation between p16INK4a ex- pression and other prognostic factors. The correlation between
Table 2. Correlation between p16INK4a expression and clini- copathological characteristics of breast cancer
p16 Total No.
Parameter p-value + (%) − (%) 50
ER+ 10 (33.3%) 20 (66.7%) 30 .154
ER− 11 (55%) 9 (45%) 20
PR+ 10 (43.5%) 13 (56.5%) 23 1
PR− 16 (59.3%) 11 (40.7%) 27
Cerb B2+ 5 (31.3%) 11 (68.3%) 16 .365 Cerb B2− 16 (47.1%) 18 (52.9%) 34
p53+ 9 (34.6%) 17 (65.4%) 26 .390
p53− 12 (50%) 12 (50%) 24
Ki-67+ 10 (47.6%) 11 (52.4%) 21 .568 Ki-67− 18 (62.1%) 11 (37.9%) 29
Grade I/II 11 (36.6%) 19 (63.4%) 30 .393 Grade III 10 (50%) 10 (50%) 20
≤2 cm 9 (40.9%) 13 (59.1%) 22 1
>2 cm 12 (42.9%) 16 (57.1%) 28
LN+ 8 (32%) 17 (68%) 25 .252
LN− 13 (52%) 12 (48%) 25
Stage I/II 20 (51.3%) 19 (48.7%) 39 .016 Stage III 1 (9.1%) 10 (90.9%) 11
≤50 7 (31.8%) 15 (68.2%) 22 .254
>50 14 (50%) 14 (50%) 28
+: positive, −: negative, ER: estrogen receptor, PR: pro- gesteron receptor, LN, lymph node
Fig. 1. Immunohistochemical detection of p16INK4a in breast cancer. A combination of nuclear and cytoplasmic staining was seen in some breast tumors (A), whereas the staining in other tumors was completely absent (B).
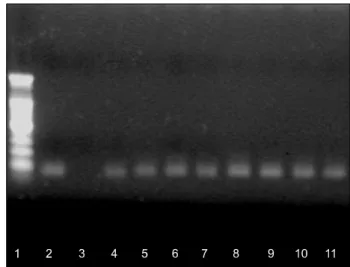
Fig. 2. Methylation-specific PCR was performed on bisulfite modified DNA obtained from tumors with loss of p16INK4a expression. DNA was amplified using methylated primers (M).
(1: DNA marker, 2: positive control, 3: negative control, 4∼11:
carcinoma tissue)
p16INK4a expression and clinicopathological features is shown in Table 2. Hypermethylation in p16INK4a was found in 36 (72%).
Twenty-three (79.3%) of 29 tumors with loss of expression of
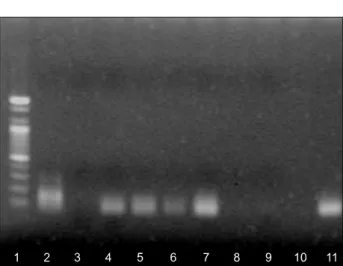
p16INK4a in immunohistochemistry showed DNA methylation of the p16INK4a gene (Fig. 2). Thirteen (61.9%) of 21 tumors with expression of p16INK4a showed DNA methylation of the p16INK4a gene (Fig. 3). The correlation between p16INK4a methy- lation and clinicopathological features is shown in Table 3.
DISCUSSION
It has been well documented that the inactivation of the p16INK4a gene is involved in disturbances of cell cycle regulation
Table 3. Correlation between p16INK4a methylation and clini- copathological characteristics of breast cancer
p16 methylation
Total No.
Parameter p-value
+ (%) − (%) 50
ER+ 23 (76.7%) 7 (23.3%) 30 .522
ER− 13 (65%) 7 (35%) 20
PR+ 18 (78.3%) 5 (21.7%) 23 .529
PR− 18 (66.7%) 9 (33.3%) 27
p16+, 2+ 13 (61.9%) 8 (38.1%) 21 .213 p16− 23 (79.3%) 6 (20.7%) 29
Cerb B2+ 13 (81.3%) 3 (18.8%) 16 .501 Cerb B2− 23 (67.6%) 11 (32.4%) 34
p53+ 20 (76.9%) 6 (23.1%) 26 .533
p53− 16 (66.7%) 8 (33.3%) 24
Ki-67+ 14 (66.7%) 7 (33.3%) 21 .534 Ki-67− 22 (75.9%) 7 (24.1%) 29
Grade I/II 24 (80%) 6 (20%) 30 .198 Grade III 12 (60%) 8 (40%) 20
≤2 cm 16 (72.7%) 6 (27.3%) 22 1
>2 cm 20 (71.4%) 8 (28.6%) 28
LN+ 20 (80%) 5 (20%) 25 .345
LN− 16 (64%) 9 (36%) 25
Stage I/II 26 (66.7%) 13 (33.3%) 39 .148 Stage III 10 (90%) 1 (10%) 11
≤50 20 (71.4%) 8 (28.6%) 28 1
>50 16 (72.7%) 6 (27.3%) 22
+: positive, −: negative, ER: estrogen receptor, PR: pro- gesteron receptor, LN: lymph node
Fig. 3. Methylation-specific PCR was performed on bisulfite modified DNA obtained from tumors with p16INK4a expression.
DNA was amplified using methylated primers (M). (1: DNA marker, 2: M positive control, 3: M negative control, 4∼11:
carcinoma tissue).
in a multitude of human malignancies. A CDK 4 inhibitor (p16INK4a) gene located at 9p21 has been shown to be inac- tivated in a variety of tumors by homozygous deletions, point mutations, or hypermethylation of its promoter region. Hyper- methylation of the p16INK4a gene has been reported to be involved in the development of a variety of human cancers.12,13) Geradts and Ingram14) found p16INK4a to be the most common target of cell cycle deregulation in invasive breast carcinomas.
Some study showed mutations or deletions of the p16INK4a gene are uncommon in breast cancer,3,4,5) and hypermethylation of the p16INK4a promoter region has been described as an alter- native mechanism to the observed lack of p16INK4a protein expression in a proportion of breast tumors.6,7) Other studies have shown that loss of p16INK4a expression in breast cancer is commonly due to deletion of the gene rather than DNA methylation of the promoter region of the gene.8,15,16) The role of DNA methylation in tumorigenesis has attracted considerable attention recently. Our study has showed a mecha- nistic correlation between DNA methylation of the p16INK4a gene and loss of p16INK4a protein as seen by immunohisto- chemistry in breast tumors. In fifty invasive breast carcinomas, we found 72% methylation in the promoter region of the p16INK4a gene. In contrast to previous studies in which 8∼60%
of analyzed breast tumors showed partial or complete methy- lation,6,7,17,18) the frequency was somewhat higher in our study.
In case p16INK4a is inactivated during tumor progression, the
frequency of methylation might depend on the average stage of tumors selected. Besides the relatively small number of tumors analyzed, another cause of difference in the stated frequency of p16INK4a methylation could be demographic diver- sity of patients.
In some human cancers, loss of p16INK4a expression, regar- dless of the mechanism, appears to confer a grave progno- sis.18,19) However, in cervical cancer, p16INK4a overexpression is related to poor prognosis.20) Both decreased and increased p16INK4a expression have been described in primary human breast cancer, and these differences in expression have not been well correlated with clinical outcome. Recently, the relationship between the expression of the p16INK4a protein and poor prognosis was reported in breast cancer.3,21,22) Our study showed that a proportion of breast cancers (58%) had loss of expression of p16INK4a and interestingly, it was related with high stage breast tumors. Twenty-three (79.3%) of 29 tumors with loss of expression of p16INK4a in immunohistochemistry
showed DNA methylation of the p16INK4a gene in our study.
Although some studies have shown that loss of p16INK4a expression in breast cancer is commonly due to hemizygous/
homozygous deletion of the gene rather than DNA methylation of the promoter region of the gene,8,15,16) our study disclosed p16INK4a promoter hypermethylation was the most important mechanism of loss of p16INK4a expression in breast cancer.
In conclusion, our study showed that loss of p16INK4a ex- pression was related with high-stage breast tumors and p16INK4a promoter hypermethylation was the main mechanism of p16INK4a protein loss. Although not an independently useful tool for the evaluation of the prognosis of the breast cancer, the expression of p16INK4a protein may be of benefit when used in combination with other molecular markers or in combination with other clinicopathological features.
REFERENCES
1) Lerma E, Esteller M, Herman JG, Prat J. Alterations of the p16/Rb/cyclin-D1 pathway in vulvar carcinoma, vulvar intrae- pithelial neoplasia, and lichen sclerosus. Hum Pathol 33, 1120-1125, 2002.
2) Jones PA, Laird PW. Cancer epigenetics comes of age. Nat Genet 21, 163-167, 1999.
3) Munot K, Bell SM, Lane S, Horgan K, Hanby AM, Speirs V. Pattern of expression of genes linked to epigenetic silencing in human breast cancer. Hum Pathol 37, 989-999, 2006.
4) Xu L, Sgroi D, Sterner CJ, Beauchamp RL, Pinney DM, Keel S, Ueki K, Rutter JL, Buckler AJ, Louis DN. Mutational analysis of CDKN2 (MTS1/p16ink4) in human breast car- cinomas. Cancer Res 4, 5262-5264, 1994.
5) Berns EM, Klijn JG, Smid M, van Staveren IL, Gruis NA, Foekens JA. Infrequent CDKN2 (MTS1/p16) gene alterations in human primary breast cancer. Br J Cancer 72, 964-967, 1995.
6) Nielsen NH, Roos G, Emdin SO, Landberg G. Methylation of the p16(Ink4a) tumor suppressor gene 5'-CpG island in breast cancer. Cancer Lett 163, 59-69, 2001.
7) Dublin EA, Patel NK, Gillett CE, Smith P, Peters G, Barnes DM. Retinoblastoma and p16 proteins in mammary carcino- ma: their relationship to cyclin D1 and histopathological parameters. Int J Cancer 79, 71-75, 1998.
8) WHO. World Health Organization: International of diseases for oncology 2nd edition. World Health Organization, Ge- neva, 1990.
9) Bloom MJC, Richardson WW. Histologic grading and prognosis in breast cancer. A study of 1709 cases. Br J Cancer Res 7, 2765-2769, 2001.
10) Frommer M, McDonald LE, Millar DS, Collis CM, Watt F, Grigg BW, Molloy PL, Paul CL. A genomic sequencing
protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc Natl Acad Sci 89, 1827-1831, 1992.
11) Clark SJ, Harrison J, Paul CL, Frommer M. High sensitivity mapping of methylated cytosines. Nucl Acid Res 22, 2290- 2297, 1994.
12) Boltze C, Zack S, Quednow C, Bettge S, Roessner A, Schneider-Stock R. Hypermethylation of the CDKN2/p16INK4A promotor in thyroid carcinogenesis. Pathol Res Pract 199, 399-404, 2003.
13) Wong YF, Chung TK, Cheung TH, Nobori T, Yu AL, Yu J, Batova A, Lai KW, Chang AM. Methylation of p16INK4A in primary gynecologic malignancy. Cancer Lett 136, 231-235, 1999.
14) Geradts J, Ingram CD. Abnormal expression of cell cycle regulatory proteins in ductal and lobular carcinomas of the breast. Mod Pathol 13, 945-953, 2000.
15) Gorgoulis VG, Koutroumbi EN, Kotsinas A, Zacharatos P, Markopoulos C, Giannikos L, Kyriakou V, Voulgaris Z, Gogas I, Kittas C. Alterations of p16-pRb pathway and chromosome locus 9p21-22 in sporadic invasive breast carcinomas. Mol Med 4, 807-822, 1998.
16) Silva J, Silva JM, Dominguez G, Garcia JM, Cantos B, Rodriguez R, Larrondo FJ, Provencio M, Espana P, Bonilla F.
Concomitant expression of p16INK4a and p14ARF in primary breast cancer and analysis of inactivation mechanisms. J Pathol 99, 289-297, 2003.
17) Di Vinci A, Perdelli L, Banelli B, et al. p16(INK4a) promoter methylation and protein expression in breast fibroadenoma and carcinoma. Int J Cancer 114, 414-421, 2005.
18) Lee MJ. Expression of G1/S phase checkpoint proteins in breast carcinoma: Relationship to clinicopathologic factors and survival rate. Cancer Res and Treat 34, 268-273, 2002.
19) Prall F, Ostwald C, Weirich V, Nizze H. p16INK4a promoter methylation and 9p21 allelic loss in colorectal carcinomas:
relation with immunohistochemical p16INK4a expression and with tumor budding. Hum Pathol 37, 578-585, 2006.
20) Kim TY, Zhao M. Aberrant cell cycle regulation in cervical carcinoma. Yonsei Med J 46, 597-613, 2005.
21) Milde-Langosch K, Bamberger AM, Methner C, Rieck G, Loning T. Expression of cell cycle-regulatory proteins RB, p16/MTS1, p27/KIP1, p21/WAF1, cyclin D1 and cyclin E in breast cancer: correlations with expression of activating protein-1 family members. Int J Cancer 87, 468-472, 2000.
22) Singh M, Parnes MB, Spoelstra N, Bleile MJ, Robinson WA.
p16 expression in sentinel nodes with metastatic breast carcinoma: evaluation of its role in developing triaging strategies for axillary node dissection and a marker of poor prognosis. Hum Pathol 35, 1524-1530, 2004.
23) Lapidus RG, Ferguson AT, Ottaviano YL, Parl FF, Smith HS, Weitzman SA, Baylin SB, Issa JP, Davidson NE. Methylation of estrogen and progesterone receptor gene 5' CpG islands correlate with lack of estrogen and progesterone receptor gene expression in breast tumors. Clin Cancer Res 2, 805-810, 1996.