CONTINUING EDUCATION COLUMN
J Korean Med Assoc 2020 March; 63(3):159-168pISSN 1975-8456 / eISSN 2093-5951 https://doi.org/10.5124/jkma.2020.63.3.159
서론
간질성 폐질환은 호흡기질환 중 드문 질환으로, 많은 임상
의사에게 낯선 질환이다. 간질성 폐질환이란 폐포와 폐포 사
이의 공간인 간질에 염증 및 섬유화가 발생하는 질환군으로,
비감염성, 비종양성 질환을 총칭한다. 따라서 간질성 폐질환
이라는 용어에는 매우 다양한 질환이 포함되어 있으며 그 질
환들의 경과는 매우 다양한데, 수년간 증상 없이 유지되는
질환부터 병이 지속적으로 진행하여 호흡부전이 발생하여
사망에까지 이를 수 있는 질환까지 이른다.
현대 고령사회로의 진행은 노인에서 발생하는 간질성 폐
질환의 발생 및 유병률을 높이고 있으며, 특히 건강검진이
매우 활발하게 시행되는 우리나라 의료시스템의 특성상, 특
별한 증상 없이 시행한 흉부 영상검사에서 우연히 초기 간질
성 폐질환이 발견되는 경우도 많아지고 있다[1,2].
그중 가장 흔하고 예후가 불량한 특발성 폐섬유증의 경
우 최근까지도 입증된 치료법이 없었기 때문에 매우 절망
스러운 불치병으로 여겨졌다. 하지만 2014년 퍼페니돈
(pirfenidone)과 닌테다닙(nintedanib)이라는 2가지 약제가
간질성 폐질환의 진단 및 치료: 특발성 폐섬유증을
중심으로 알아보기
강 혜 린·최 선 미 서울대학교병원 호흡기내과Diagnosis and treatment of interstitial lung disease:
focusing on idiopathic pulmonary fibrosis
Hye-Rin Kang, MD·Sun Mi Choi, MD
Division of Pulmonary and Critical Care Medicine, Department of Internal Medicine, Seoul National University Hospital, Seoul, Korea
Received: February 7, 2020 Accepted: March 3, 2020 Corresponding author: Sun Mi Choi
E-mail: [email protected] © Korean Medical Association
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons. org/licenses/by-nc/3.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Interstitial lung disease (ILD) is a rare condition characterized by extensive inflammation and fibrosis mainly involving the pulmonary interstitium or alveoli. Usually, patients with ILD clinically present with chronic cough and exertional dyspnea. ILD is classified into subtypes based on clinical characteristics, detailed history obtained from patients, and radiological, and/or histopathological features. The most common type of idiopathic interstitial pneumonia is idiopathic pulmonary fibrosis (IPF). IPF is a chronic progressive fibrosing ILD and is associated with poor prognosis. An exclusive diagnosis of IPF requires no known condition causing ILD and typical radiological and/or histopathological features of lung fibrosis. Fibrosis observed in this condition is attributable to repetitive epithelial injury with consequent abnormal wound healing in genetically susceptible and elderly individuals. Currently, pirfenidone and nintedanib are useful disease-modifying agents available to treat IPF. In this article, we review the concept, diagnosis, clinical course, and treatment of ILD.
특발성 폐섬유증의 진행속도를 늦춰준다는 연구결과가 발표
됨에 따라 이 질환의 치료에 새로운 장이 열리게 되었다. 국
내에서 역시 이러한 약제의 사용이 대학병원에 국한되지 않
고 점차 그 사용이 넓어지고 있다.
이번 리뷰에서는 간질성 폐질환에 대한 간단한 소개와 분
류, 임상양상 및 진단과정에 대해서 살펴보고, 특히 특발성
간질성 폐렴 중 가장 흔한 특발성 폐섬유증에 대해서 좀더
자세히 정리해보고자 한다.
간질성 폐질환의 정의 및 역학
간질성 폐질환은 폐 간질의 세포 증식, 염증세포 침투 혹
은 폐 실질의 섬유화를 특징으로 하는 비종양성, 비감염성
질환을 이르는 용어로[3] 다양한 폐질환을 포함하고 있는
광범위한 질환군이며, 약 200개가 넘는 진단이 포함된다.
전체 간질성 폐질환의 유병률은 10만 명당 67-81명으로 추
정하고 있으며 발생률은 10만 명당 26-32명으로 추정한다
[4,5].
간질성 폐질환의 분류는 다양한데, 그 중 임상적으로 중요
한 분류는 간질성 폐질환을 유발할 수있다고 알려진 원인들
즉 결체조직질환, 약물 혹은 치료, 직업 및 환경노출 등과 관
련된 간질성 폐질환들, 그리고 이러한 알려진 유발원인을 찾
을 수 없는 특발성 간질성 폐렴, 그리고 그외 랑게르한스 폐
조직구증이나 사르코이드증과 같이 다양한 일차성 간질성
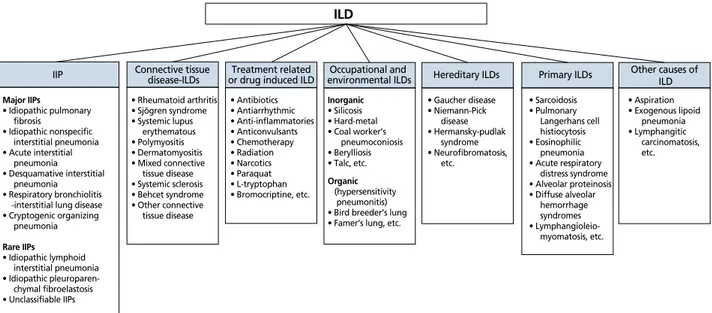
폐질환 등으로 나눌 수 있다(Figure 1) [6-9].
진단적 접근법
간질성 폐질환은 앞서 설명한 바와 같이 매우 다양한 질환
을 포함하며 질환에 따라 그 예후 및 치료가 다르기 때문에
간질성 폐질환 중 어떠한 질환인지를 정확하게 진단하는 것
이 매우 중요하다. 이러한 간질성 폐질환의 세부 진단은 임
상적 병력 및 신체검진 소견, 영상학적, 병리학적 소견을 종
합해서 이루어지게 되며, 이를 위해서 호흡기내과, 영상의학
과, 병리과 등 여러 과의 임상 의사가 함께 상의하는 다학제
적 토의 과정이 매우 중요하다[10].
가장 먼저 환자의 병력에 대한 자세한 확인이 필요하며,
증상의 시작 및 진행 속도, 직업력, 투약력, 흡연력, 석면 및
유해물질의 환경 노출력과 가족력 및 여행력 등을 확인해야
한다. 또한 간질성 폐질환의 중요한 유발원인인 결체조직질
환을 포함한 과거 병력을 확인해야 한다. 이전에 진단된 결
체조직질환이 없다고 하더라도, 관절통, 안구 혹은 구강 건
IIP Hereditary ILDs
Major IIPs • Idiopathic pulmonary fibrosis • Idiopathic nonspecific interstitial pneumonia • Acute interstitial pneumonia • Desquamative interstitial pneumonia • Respiratory bronchiolitis -interstitial lung disease • Cryptogenic organizing pneumonia Rare IIPs • Idiopathic lymphoid interstitial pneumonia • Idiopathic chymal fibroelastosis • Unclassifiable IIPs • Rheumatoid arthritis • Sjögren syndrome • Systemic lupus erythematous • Polymyositis • Dermatomyositis • Mixed connective tissue disease • Systemic sclerosis • Behcet syndrome • Other connective tissue disease Connective tissue disease-ILDs • Antibiotics • Antiarrhythmic • Anti-inflammatories • Anticonvulsants • Chemotherapy • Radiation • Narcotics • Paraquat • L-tryptophan • Bromocriptine, etc. Inorganic • Silicosis • Hard-metal • Coal worker’s pneumoconiosis • Berylliosis • Talc, etc. Organic (hypersensitivity pneumonitis) • Bird breeder’s lung • Famer’s lung, etc.
• Gaucher disease • Niemann-Pick disease • Hermansky-pudlak syndrome • Neurofibromatosis, etc. Primary ILDs • Sarcoidosis • Pulmonary Langerhans cell histiocytosis • Eosinophilic pneumonia • Acute respiratory distress syndrome • Alveolar proteinosis • Diffuse alveolar hemorrhage syndromes • myomatosis, etc. Other causes of ILD • Aspiration • Exogenous lipoid pneumonia • Lymphangitic carcinomatosis, etc. Treatment related
or drug induced ILD environmental ILDsOccupational and
ILD
Kang HR·Choi SM·Interstitial lung disease
조, 피부발진이나 근육의 위약감, 근육통 등의 전신증상에
대한 꼼꼼한 병력청취가 필요하다. 위식도 역류증 및 반복적
인 흡인의 유무도 확인해보아야 한다. 청진 시 주로 양측 하
부 폐에서 흡기 시 수포음이 들릴 수 있으며, 곤봉지도 관찰
될 수 있다.
간질성 폐질환의 진단에 있어 가장 중요한 검사는 흉부 고
해상도 단층촬영(high-resolution computed tomography,
HRCT)으로, 흉부 X선 촬영에서 간질성 폐질환이 의심되거
나 임상적으로 간질성 폐질환이 의심되는 모든 환자에서 필
수적으로 시행되어야 한다. 적절한 임상적 상황에서 HRCT
상 질병 특징적인 영상의학적 소견이 있다면 폐생검을 하지
않아도 간질성 폐질환 중 어떠한 병인지 진단을 할 수 있는
경우도 있다.
간질성 폐질환이 의심되는 환자에게는 폐활량 검사 및 폐
확산능 검사를 시행하게 되는데, 대부분의 간질성 폐질환에
서는 노력성 폐활량(forced vital capacity, FVC) 및 폐확산
능(diffusing capacity for carbon monoxide, DLCO)이 저
하되어 있다. 이 검사들은 질병의 진행을 평가하기에 유용하
기 때문에 정기적으로 시행하게 된다.
호산구 폐렴이나 사르코이드증(sarcoidosis) 같은 일부 간
질성 폐질환에서는 기관지내시경을 통한 기관지폐포세척술
이 진단에 도움이 될 수 있으며[11], 림프관평활근종증과 같
은 일부 질환에서는 기관지내시경을 통한 경기관지 폐생검
이 도움이 되기도 한다. 그러나 조직검사가 필요한 대부분의
간질성 폐질환의 경우 경기관지 폐생검으로는 정확한 진단
을 하기가 어렵기 때문에 흉강경을 통한 폐생검이 추천된다.
환자의 자세한 병력과 영상 및 병리 검사결과를 바탕으로 한
다학제 논의를 통해 간질성 폐질환 중 어떠한 질환인지 진단
을 내리게 된다[8].
치료적 접근법 및 예후
간질성 폐질환은 그 종류에 따라 치료방침이 달라지는데,
특정 원인에 의해 유발된 간질성 폐질환의 경우 그 원인을
제거 혹은 치료하는 것이 중요하다. 즉 결제조직질환과 관련
된 간질성 폐질환에서는 스테로이드와 다양한 면역억제제가
사용되며, 환경 혹은 직업적 노출과 관련된 간질성 폐질환에
서는 이러한 노출을 중단하는 것이 필수적이다. 원인을 찾을
수 없는 특발성 폐렴 중 기질화 폐렴이나 비특이성 간질성
폐렴은 스테로이드 치료가 비교적 좋은 효과를 보이며, 특발
성 폐섬유증의 경우 스테로이드 치료는 오히려 환자에게 해
가 될 수 있고 항섬유약제가 사용된다. 최근 이러한 항섬유
약제가 특발성 폐섬유증 이외의 다양한 진행성 폐섬유증 환
자에서도 그 효과가 입증되고 있어, 앞으로 그 적용증이 넓
어질 것으로 생각되고 있다[12].
예후 역시 질환에 따라서 매우 다양한데, 특별한 치료 없
이 저절로 호전되거나 안정적으로 유지되는 질환부터 매우
급성 경과를 보여 사망에 이르는 질환까지 다양한 경과를 보
인다.
특발성 폐섬유증
1. 역학
특발성 폐섬유증은 특발성 간질성 폐렴 중에서 가장 흔하
며, 가장 활발히 연구가 진행되고 있는 병이다. 유병률 및 발
병률은 국가에 따라서 다양하나, 북미 및 유럽에 비해서 아
시아인의 유병률은 낮은 것으로 알려져 있고 북미에서는 10
만 명당 3-9명 정도의 발병률, 유병률은 10만 명당 10-60
명 가량이나 국내에서는 발병률이 10만 명당 1.7명으로 비
교적 적은 편이다[13]. 북미에서 특발성 폐섬유증의 유병률
은 10년 전에 비해서 2배 가량 증가되고, 입원 및 사망이 늘
고 있으며 사회 전반적인 질병의 부담이 늘어나고 있다. 특
발성 폐섬유증은 남성에서 더 흔하고 대부분 50대 이상에서
발생한다[7]. 하지만 가족성 폐섬유증 및 일부 환자에서는
50대 이하에서도 발생할 수 있다[3].
2. 위험인자 및 발병기전
잘 알려져 있는 위험인자로는 남성, 70대 이상의 고령, 흡
연이 있고 그외에 역류성 식도염, 분진과 관련된 직업력, 대
기오염 등이 위험인자로 알려져 있다[7,14,15]. 그중 고령은
특별성 폐섬유증의 발병에 있어서 가장 강한 위험인자라고
알려져 있다[16].
예전에는 특발성 폐섬유증의 발병기전이 만성염증으로 인
한 섬유화라고 생각되었으나, 여러 연구결과를 바탕으로 현
재는 질병 감수성이 있는 개인에서 반복적인 상피세포의 손
상으로 비정상적인 상처회복 과정이 유발되어 과도한 섬유
아세포의 증식 및 기질 생성에 의한 폐 실질의 섬유화가 진
행되어 발생한다고 생각되고 있다[17,18].
이러한 발병기전을 좀더 자세히 살펴보면, 고령 또는 유전
적인 선행 요인을 가진 환자들이 흡연이나 분진 등의 공해물
질, 바이러스 감염, 담배연기, 위식도 역류 등으로 인한 물리
적 자극, 그 외 손상을 야기시킬 수 있는 다양한 환경적 요인
에 지속적으로 노출되게 되면, 텔로미어의 기능저하, 미토콘
드리아 기능이상 등의 후생 유전적 이상으로 인해서 폐포상
피세포의 상처치유 경로가 비정상적으로 활성화되고 비정상
적 손상 치유반응을 나타내게 된다[16]. 특히 상피세포 중 폐
포 표면활성제를 분비하고 1형 폐포상피세포의 재생을 관리
하는 것으로 알려진 2형 폐포상피세포의 이동 및 증식이 유
발되며, 이는 표피전구세포를 증가시키고, 비정상적인 폐포
의 재생산이 일어나게 된다[19].
손상된 상피세포는 섬유화 매개물질 및 다양한 성장인자
들을 분비하여 섬유아세포를 모으게 된다. 모여든 섬유아세
포는 알파평활근액틴(α-smooth muscle actin)을 발현하
는 근섬유아세포로 분화하게 된다. 근섬유아세포는 아교질
을 분비하는데, 이 때 근섬유아세포의 아교질 분비를 촉진
시키는 자극신호와 억제신호의 불균형이 발생하면 폐 실질
내에 과다 분비된 아교질이 축적되게 된다[17]. 또한 폐포의
포식세포와 같은 선천면역세포들도 폐 섬유화에 기여한다
[7,20]. 이러한 과정을 통해 폐포와 모세혈관 사이의 간질에
섬유화가 진행되면서 결국에는 산소 교환능이 감소하게 된
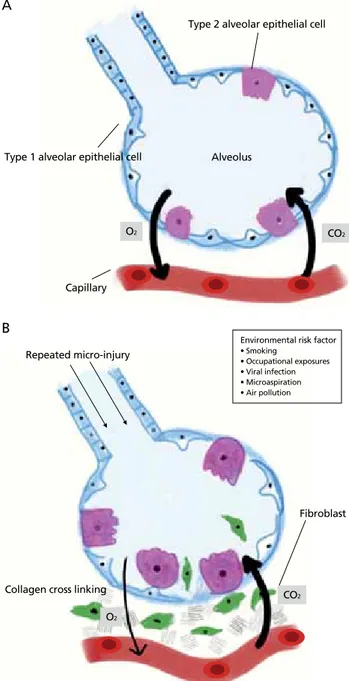
다(Figure 2) [19].
특발성 폐섬유증과 연관이 있는 유전자들에 대한 연구도
활발하게 진행되고 있는데, 환자의 방어능력과 연관이 있
는 MUC5B, ATP11A, TOLLIP, 텔로미어 유지와 관련 있는
TERT, TERC, 상피세포의 방어 기능과 관련이 있는 DSP,
DPP9 등의 유전자 이상이 질환의 발병과 연관이 있다고 알
려져 있다[7,18].
3. 증상 및 임상양상
주로 50세 이후에 발병하며, 서서히 진행하는 가래를 동반
하지 않은 기침 및 운동 시 호흡곤란과 피로함이 주된 증상
이다[19]. 초기에는 수개월에 걸쳐서 천천히 호흡곤란이 진행
하는 양상을 보이므로, 만성 호흡곤란을 일으킬 수 있는 다
른 질환(심부전 및 만성 폐쇄성 폐질환 등)과의 감별이 필요
Figure 2. Pathophysiology of idiopathic pulmonary fibrosis (IPF). (A) Normal lung and (B) IPF lung. Drawn by authors.
Type 2 alveolar epithelial cell
Type 1 alveolar epithelial cell
Capillary
Repeated micro-injury
Environmental risk factor • Smoking • Occupational exposures • Viral infection • Microaspiration • Air pollution Fibroblast
Collagen cross linking Alveolus CO2 CO2 O2 O2 B
Type 2 alveolar epithelial cell
Type 1 alveolar epithelial cell
Capillary
• Viral infection • Microaspiration • Air pollution
Fibroblast
Collagen cross linking Alveolus CO2 CO2 O2 O2 A
Kang HR·Choi SM·Interstitial lung disease
하다. 진찰 소견 상 양쪽 폐 하부에 흡기성 수포음이 들리며
25-50%의 환자에서는 곤봉지를 보일 수 있다[18]. 만성적인
호흡곤란에 비해서 적혈구 증다증은 거의 없으며 폐기능 검
사상 제한적 폐기능 장애 소견이 확인되고 DLCO의 감소가
관찰된다. 특발성 폐섬유증의 초기에는 FVC 및 DLCO가 정
상으로 유지되기도 하지만, 병이 진행할수록 FVC 및 DLCO
가 감소되며 산소포화도 특히 운동 시 산소포화도가 감소한
다. 초기에는 흉부 X선 검사에서 정상으로 보일 수 있으나 병
이 진행할수록 양측 하부, 흉막하부터 진행하는 망상형 침윤
이 보이게 된다.
4. 진단
환자가 만성적인 기침 및 운동 시 호흡곤란이 있고 흡기
시 수포음이 청진된다면 간질성 폐질환의 가능성을 고려해
야 한다. 앞서 설명한 바와 같이 간질성 폐질환이 의심되는
경우 알려진 원인을 확인하기 위한 자세한 병력청취가 필수
적이며, 특히 관절, 근육, 피부 등의 이
상 소견을 확인하여 결체조직질환의 가
능성을 확인해야 한다[18]. 의심이 되는
결체조직질환이 있다면 류마티스인자
나 항핵항체 등 자가항체 검사를 시행
하고 류마티스내과 전문의에게 의뢰하
여 결체조직질환의 유무를 확인하는 것
이 필요하다. 병력청취 및 검사상 알려
진 원인을 찾을 수 없을 경우 특발성 간
질성 폐렴이라고 할 수 있다. 간질성 폐
질환이 의심되는 모든 환자에서 HRCT
를 촬영해야 하는데, 1.25 mm 미만으
로 재건되는 프로토콜로 촬영하는 것이
추천되며, 가능하다면 흡기와 호기 시
각각 컴퓨터단층촬영(computed
tomo-graphy, CT)을 하는 것이 감별진단에
도움이 된다.
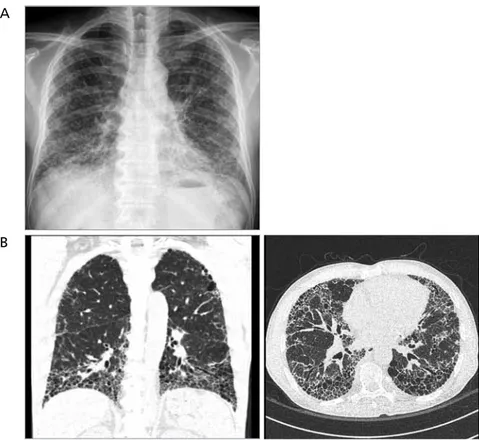
특발성 폐섬유증의 전형적인 HRCT
소견은 주로 폐의 기저부, 흉막 아래에
망상 음영 및 견인성 기관지 확장증, 벌
집모양의 폐 소견이 관찰되면서 이 외의 다른 질환을 시
사하는 소견(섬유화 병변보다 더 넓은 간유리 음영, 폐경
결, 중-상엽 침범 등)이 없어야 한다. 이러한 소견을 영상
의학적 usual interstitial pneumonia pattern (UIP형)이라
고 한다(Figure 3). 예전에는 특발성 폐섬유증을 진단하기
위해서는 조직검사를 시행하여 조직학적 UIP형을 확인하는
것이 필수적이었으나, 흉부 CT에서 전형적인 UIP 소견이
보일 경우 병리검사 상 UIP형과 일치도가 매우 높다는 것이
밝혀지면서[21], 특발성 간질성 폐렴 환자의 HRCT상 전형
적인 UIP형이 관찰되는 경우 조직검사 없이 특발성 폐섬유
증을 확진할 수 있다[22].
영상 검사에서 전형적인 UIP형이 아닐 경우 폐 조직검사
가 필요하며, 기관지 내시경으로시행하는 경기관지 조직검
사의 경우 정확한 진단이 어렵기 때문에 수술적 폐생검, 주
로 흉강경을 통한 폐생검을 시행한다. 병리학적으로 정상 폐
조직, 간질의 염증, 섬유아세포병소, 진한 아교질 섬유증, 벌
Figure 3. Typical usual interstitial pneumonia pattern of chest X-ray and high-resolution computed tomography of patient with idiopathic pulmonary fibrosis. Increased extent of diffuse subpleural reticular opacities, traction bronchiectasis and honeycombing in both lungs showing a basilar predominancy, consistent with usual interstitial pneumonia pattern. (A) Chest posteroanterior and (B) high-resolution computed tomography. The patient provided written informed consent for the publication.
B A
집모양 변화 등의 여러 소견이 혼재되어 나타나는 일시적 이
질성이 특징적이다. 그러나 UIP형과 비슷한 섬유화 소견은
만성 과민성 폐렴이나 말기의 사르코이드증 등 다른 질환에
서도 만성적인 말기로 진행하면 보일 수 있는 소견이기 때문
에 조직에서 다른 폐질환을 시사하는 소견은 없는지 확인해
야 한다.
위와 같이 증상, 병력, 신체검진 및 혈액검사 등의 임상 소
견과 HRCT 소견, 그리고 조직검사결과를 바탕으로 경험이
풍부한 호흡기내과 및 병리과, 영상의학과 의사의 다학제적
토의를 통해 간질성 폐질환 중 어떠한 질환인지 진단하게 된
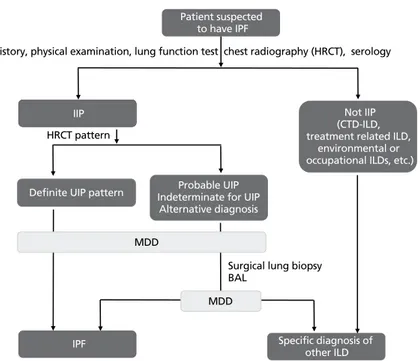
다(Figure 4) [23].
5. 치료
특발성 폐섬유증은 이전에는 질병의 경과를 바꾸는 입증
된 약물 치료가 없었으나, 2014년에 퍼페니돈과 닌테다닙
이라는 약제가 개발되면서 치료에 새로운 지평이 열리게 되
었다. 이 두 가지 약은 무작위배정 위약대조군연구에서 1
년 동안 FVC의 감소속도를 50% 정도 늦추었으며, 급성악화
도 감소시켰다[24-27]. 이에 두 가지 약제 모두 2014년 미
국 식약청에서 특발성 폐섬유증의 치료
제로 승인되었다. 이후 시행된 메타분석
연구에서는 사망률을 줄이는 효과도 보
여주었다[28].
이 두 약물의 기전을 살펴보면, 닌
테다닙은 타이로신 인산화효소 억제제
(tyrosine kinase inhibitor)로 혈소판
유래 성장인자 수용체, 혈관내피세포
성장인자 수용체 및 섬유아세포 성장인
자 수용체에 작용하여 성장인자경로를
억제하는 작용을 한다. 퍼페니돈은 항
염증 및 항섬유화 작용을 하는 약으로
콜라겐합성 및 베타종양성장인자와 알
파종양괴사인자 및 섬유아세포의 증식
을 억제한다. 현재 우리나라에서는 닌
테다닙은 급여 적용이 되지 않으며 퍼
페니돈만 급여 적용이 되기 때문에 대
부분의 환자에서 퍼페니돈을 사용하고 있다. 퍼페니돈은 처
음에는 한번 복용 시 200 mg씩 하루 3회로 복용을 시작하
여 약 2주 간격으로 400 mg 3회, 600 mg 3회까지 증량한
다. 흔한 부작용으로는 식욕감소, 위약감 및 소화기계 부작
용이 있으며, 소화불량, 구역, 구토가 발생할 수 있고 간독
성이 있을 수 있기 때문에 간수치 모니터링이 지속적으로
필요하다. 또한 광민감성 발진이 발생할 수 있어서 자외선
차단제 도포 및 자외선 노출을 줄이도록 교육해야 한다. 최
근 퍼페니돈의 국내 보험기준이 좀더 넓게 확장되면서 현재
는 특발성 폐섬유증 환자에서 FVC가 90% 이하 혹은 폐확
산능이 80% 이하일 때 급여 적용이 되며, 그 기준에 맞지
않더라도 폐기능 저하(연간 FVC 예측치 감소량이 10% 이
상이거나 연간 FVC 예측치 200 mL 이상 감소), 임상증상
악화, 흉부영상 악화 소견 중에 두 가지 이상 만족 할 때는
급여 적용이 되어 퍼페니돈을 사용할 수 있다.
항섬유약제 투약을 언제 시작할 것인지에 대해서는 아직
까지 논란이 있다. 폐기능이 보존된 초기에 항섬유약제를 투
약하였을 때도 병의 진행을 늦추는 효과는 동일하다는 연구
결과가 발표됨에 따라 증상이 없더라도 조기치료를 지지하
Figure 4. Diagnostic algorithm for idiopathic pulmonary fibrosis (IPF). HRCT, high-resolution computed to-mography; IIP, idiopathic interstitial pneumonia; CTD, connective tissue disease; ILD, interstitial lung disease; UIP, usual interstitial pneumonia; BAL, bronchoalveolar lavage; MDD, multidisciplinary discussion.
Patient suspected to have IPF
IIP
IPF
Definite UIP pattern Indeterminate for UIPProbable UIP Alternative diagnosis
Specific diagnosis of other ILD
Not IIP (CTD-ILD, treatment related ILD,
environmental or occupational ILDs, etc.)
History, physical examination, lung function test chest radiography (HRCT), serology
MDD
Surgical lung biopsy BAL
HRCT pattern
Kang HR·Choi SM·Interstitial lung disease
는 의사들도 있다. 그러나 항섬유약제의 입증된 효과가 질환
의 호전이라기 보다는 질환의 진행을 늦추는 것이라는 점과,
환자마다 질환의 진행속도가 다르다는 점을 고려할 때, 폐기
능이 안정적으로 유지되고 증상이 없는 일부 환자에서는 폐
기능 감소 경향을 면밀하게 관찰하면서 약물 시작시기를 결
정해 볼 수 있겠다. 특히 고령에서는 퍼페니돈의 위장관 부
작용과 식욕의 저하로 인해 전신위약감 등이 발생하면서 전
신상태가 오히려 악화될 수도 있으므로, 특발성 폐섬유증의
적절한 치료를 위해서는 경험이 많은 의사에게 진료의뢰를
하는 것이 권고된다[29].
비약물적 치료로는 저산소증이 있는 경우 산소공급이 필
요하며, 모든 환자에서 호흡재활이 추천된다. 약물치료에도
불구하고 질환이 진행될 경우에는 폐이식만이 그 생존 기간
을 연장시킬 수 있다.
6. 특발성 폐섬유증의 급성악화
특발성 폐섬유증의 급성악화는 임상적으로 심각한 호흡부
전을 일으키는 광범위한 폐포의 이상으로 정의할 수 있다.
일반적으로 한달 이내에 시작된 급성호흡악화로 나타나며,
기흉, 흉수, 폐색전증 등과 같은 폐실질 외의 원인이 배제되
고, 체액과다 및 심부전 없이 흉부 CT상 양측의 미만성 간
유리음영이 관찰되는 경우 특발성 폐섬유증의 급성악화라고
진단할 수 있다. 이런 급성악화는 특정 원인으로 유발된 급
성악화와 특발성 급성악화로 분류할 수 있으며 흔한 급성악
화의 원인으로는 감염, 수술이나 시술, 약제 독성, 흡인 등
이 있다[30].
급성악화의 발생률은 보고에 따라 다양하다. 6개의 임상
시험을 메타분석하여 2014년에 발표한 연구에서는 100인년
당 4.1의 급성악화 발생이 보고되었고[31], 코호트연구들에
서 보고된 발생률은 일반적으로 이보다 높아 10% 전후로 보
고되고 있다. 특히 2011년 국내 환자들을 분석하여 발표한
연구에서는 1년 및 3년 발생률이 14.2%, 20.7%로 보고되었
다[32]. 최근 급성악화의 진단기준이 좀더 완화된 것을 고려
할 때 국내 특발성 폐섬유증 환자에서 급성악화의 발생은 이
보다 높을 것으로 생각된다.
급성악화의 사망률은 약 50%로 알려져 있고, 급성악화
이후 중간 생존율은 3-4개월로 그 예후가 매우 불량하다
[30,33]. 급성악화의 발생 위험인자로는 진단 시 낮은 폐기
능, 6개월간 노력성 폐활량의 감소정도, 낮은 산소포화도,
폐고혈압, 호흡곤란의 중증도 및 흉부 CT상 섬유화의 범위
등이 있으며 보통은 질환이 진행된 상태일수록에 취약하다
고 알려져 있다. 그 외에 각종 검사 및 수술 후에 급성악화가
발생할 수 있다[30].
급성악화의 치료에 대해서는 아직까지 근거가 충분히 정
립되어 있지는 않지만 국제가이드라인에서는 경험적으로 고
용량의 코르티코스테로이드를 투약하는 것을 추천하고 있다
[34]. 급성악화 환자에서의 기관 삽관 및 기계 호흡에 대해서
는 논란의 여지가 있는데, 폐이식을 고려하는 경우에는 가교
치료로 기계호흡을 적용해볼 수 있겠다[30].
7. 예후
특발성 폐섬유증은 예후가 나쁜 질환으로, 진단 이후 중
앙생존기간이 3-4년으로 짧다[8,19,35]. 그러나 질병의 경
과는 환자에 따라 다양하게 나타날 수 있으며 매우 천천히
진행하거나 매우 빠르게 진행하는 경우도 있어 환자 개개인
의 경과는 예측하기가 어렵다[18]. 대부분의 환자들은 질병
자체의 진행 및 만성적인 호흡부전으로 사망하게 되지만, 급
성악화가 발생할 경우 사망 혹은 폐기능 악화로 인해 생존기
간의 단축을 유발할 수 있다[36].
환자의 호흡곤란이 심할수록, 폐확산능 악화 및 6분 도보
검사상 산소포화도가 떨어질수록, 노력성 폐활량이 6-12개
월 사이에 10% 이상 감소할 때, CT상의 벌집모양의 섬유화
가 증가되거나 폐고혈압이 발생하게 되는 경우에는 나쁜 예
후를 보인다고 알려져 있다[29,37,38]. 그 외에도 폐색전증
및 폐암, 폐고혈압 등이 병발되어 예후를 악화시킬 수 있다.
특히 특발성 폐섬유증 환자에서는 폐암의 위험이 6-20%까
지 올라가기 때문에, 면밀한 관찰이 요구된다[39].
결론
간질성 폐질환은 그 종류가 다양하고 질환에 따른 치료법
과 예후가 각각 다른 병으로, 최근 CT의 시행이 증가함에 따
라 일선 진료 현장에서 경험하게 되는 경우가 늘어나고 있
다. 원인이 분명하지 않은 장기간의 기침과 운동시 호흡곤
란, 흡기 시 수포음과 흉부엑스선상 양측 기저부 폐의 망상
형 소견이 보인다면 반드시 간질성 폐질환을 의심해 보아야
한다. 간질성 폐질환의 정확한 진단에는 꼼꼼한 병력청취가
중요하며, 혈액검사, HRCT 및 조직검사까지 필요할 수 있
다. 이러한 검사결과를 바탕으로 다학제적 토론을 통한 진단
이 이루어지는 만큼 임상, 병리, 영상정보를 통합할 수 있는
전문가 집단으로 진료의뢰를 하여 정확한 진단이 이루어지
게 하는 것이 중요하겠다.
특발성 간질성 폐렴 중에서 가장 흔한 특발성 폐섬유증은
3-4년 이내에 사망에 이를 수 있는 예후가 매우 불량한 질
환으로, 최근 개발된 퍼페니돈과 닌테다닙이라는 약제가 병
의 진행을 늦출 수 있어 빠른 진단 및 치료가 환자의 예후 향
상에 기여할 것으로 예상한다.
찾아보기말:
간질성 폐질환; 특발성 간질성 폐렴; 특발성 폐섬유증ORCID
Hye-Rin Kang, https://orcid.org/0000-0002-8852-8736 Sun Mi Choi, https://orcid.org/0000-0002-0742-6085
Conflict of Interest
No potential conflict of interest relevant to this article was reported.
References
1. Flaherty KR, Brown KK, Wells AU, Clerisme-Beaty E, Collard HR, Cottin V, Devaraj A, Inoue Y, Le Maulf F, Richeldi L, Schmidt H, Walsh S, Mezzanotte W, Schlenker-Herceg R. Design of the PF-ILD trial: a double-blind, randomised, placebo-controlled phase III trial of nintedanib in patients with progressive fibrosing interstitial lung disease. BMJ Open Respir Res 2017;4:e000212.
2. Jin GY, Lynch D, Chawla A, Garg K, Tammemagi MC, Sahin H, Misumi S, Kwon KS. Interstitial lung abnormalities in a CT
lung cancer screening population: prevalence and progression rate. Radiology 2013;268:563-571.
3. Rosas IO, Dellaripa PF, Lederer DJ, Khanna D, Young LR, Martinez FJ. Interstitial lung disease: NHLBI Workshop on the primary prevention of chronic lung diseases. Ann Am Thorac Soc 2014;11 Suppl 3:S169-S177.
4. Coultas DB, Zumwalt RE, Black WC, Sobonya RE. The epide-miology of interstitial lung diseases. Am J Respir Crit Care Med 1994;150:967-972.
5. Demedts M, Wells AU, Antó JM, Costabel U, Hubbard R, Cullinan P, Slabbynck H, Rizzato G, Poletti V, Verbeken EK, Thomeer MJ, Kokkarinen J, Dalphin JC, Taylor AN. Interstitial lung diseases: an epidemiological overview. Eur Respir J Suppl 2001;32:2s-16s.
6. Stoller JK. Murray & Nadelʼs Textbook of Respiratory Medi-cine, 6th Edition. Ann Am Thorac Soc 2015;12:1257-1258. 7. Lederer DJ, Martinez FJ. Idiopathic pulmonary fibrosis. N
Engl J Med 2018;378:1811-1823.
8. Cottin V, Hirani NA, Hotchkin DL, Nambiar AM, Ogura T, Otaola M, Skowasch D, Park JS, Poonyagariyagorn HK, Wuyts W, Wells AU. Presentation, diagnosis and clinical course of the spectrum of progressive-fibrosing interstitial lung diseases. Eur Respir Rev 2018;27:180076.
9. Travis WD, Costabel U, Hansell DM, King TE Jr, Lynch DA, Nicholson AG, Ryerson CJ, Ryu JH, Selman M, Wells AU, Behr J, Bouros D, Brown KK, Colby TV, Collard HR, Cordeiro CR, Cottin V, Crestani B, Drent M, Dudden RF, Egan J, Flaherty K, Hogaboam C, Inoue Y, Johkoh T, Kim DS, Kitaichi M, Loyd J, Martinez FJ, Myers J, Protzko S, Raghu G, Richeldi L, Sverzellati N, Swigris J, Valeyre D; ATS/ERS Committee on Idiopathic Interstitial Pneumonias. An official American Thoracic Society/European Respiratory Society statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med 2013;188:733-748.
10. De Sadeleer LJ, Meert C, Yserbyt J, Slabbynck H, Verschakelen JA, Verbeken EK, Weynand B, De Langhe E, Lenaerts JL, Nemery B, Van Raemdonck D, Verleden GM, Wells AU, Wuyts WA. Diagnostic ability of a dynamic multidisciplinary discussion in interstitial lung diseases: a retrospective obser-vational study of 938 cases. Chest 2018;153:1416-1423. 11. Meyer KC, Raghu G, Baughman RP, Brown KK, Costabel U,
du Bois RM, Drent M, Haslam PL, Kim DS, Nagai S, Rottoli P, Saltini C, Selman M, Strange C, Wood B; American Thoracic Society Committee on BAL in Interstitial Lung Disease. An official American Thoracic Society clinical practice guideline: the clinical utility of bronchoalveolar lavage cellular analysis in interstitial lung disease. Am J Respir Crit Care Med 2012; 185:1004-1014.
12. Flaherty KR, Wells AU, Cottin V, Devaraj A, Walsh SLF, Inoue Y, Richeldi L, Kolb M, Tetzlaff K, Stowasser S, Coeck C, Clerisme-Beaty E, Rosenstock B, Quaresma M, Haeufel
Kang HR·Choi SM·Interstitial lung disease
T, Goeldner RG, Schlenker-Herceg R, Brown KK; INBUILD Trial Investigators. Nintedanib in progressive fibrosing inter-stitial lung diseases. N Engl J Med 2019;381:1718-1727. 13. Gjonbrataj J, Choi WI, Bahn YE, Rho BH, Lee JJ, Lee CW.
Incidence of idiopathic pulmonary fibrosis in Korea based on the 2011 ATS/ERS/JRS/ALAT statement. Int J Tuberc Lung Dis 2015;19:742-746.
14. Awadalla NJ, Hegazy A, Elmetwally RA, Wahby I. Occupa-tional and environmental risk factors for idiopathic pulmo-nary fibrosis in Egypt: a multicenter case-control study. Int J Occup Environ Med 2012;3:107-116.
15. Sgalla G, Iovene B, Calvello M, Ori M, Varone F, Richeldi L. Idiopathic pulmonary fibrosis: pathogenesis and manage-ment. Respir Res 2018;19:32.
16. Wolters PJ, Collard HR, Jones KD. Pathogenesis of idiopathic pulmonary fibrosis. Annu Rev Pathol 2014;9:157-179. 17. Selman M, King TE, Pardo A; American Thoracic Society;
European Respiratory Society; American College of Chest Physicians. Idiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann Intern Med 2001;134:136-151.
18. Richeldi L, Collard HR, Jones MG. Idiopathic pulmonary fibrosis. Lancet 2017;389:1941-1952.
19. Martinez FJ, Collard HR, Pardo A, Raghu G, Richeldi L, Selman M, Swigris JJ, Taniguchi H, Wells AU. Idiopathic pulmonary fibrosis. Nat Rev Dis Primers 2017;3:17074. 20. Bagnato G, Harari S. Cellular interactions in the pathogenesis
of interstitial lung diseases. Eur Respir Rev 2015;24:102-114. 21. Raghu G, Lynch D, Godwin JD, Webb R, Colby TV, Leslie KO,
Behr J, Brown KK, Egan JJ, Flaherty KR, Martinez FJ, Wells AU, Shao L, Zhou H, Pedersen PS, Sood R, Montgomery AB, O'Riordan TG. Diagnosis of idiopathic pulmonary fibrosis with high-resolution CT in patients with little or no radiological evidence of honeycombing: secondary analysis of a randomised, controlled trial. Lancet Respir Med 2014;2:277-284.
22. Nishimura K, Kitaichi M, Izumi T, Nagai S, Kanaoka M, Itoh H. Usual interstitial pneumonia: histologic correlation with high-resolution CT. Radiology 1992;182:337-342.
23. Raghu G, Remy-Jardin M, Myers JL, Richeldi L, Ryerson CJ, Lederer DJ, Behr J, Cottin V, Danoff SK, Morell F, Flaherty KR, Wells A, Martinez FJ, Azuma A, Bice TJ, Bouros D, Brown KK, Collard HR, Duggal A, Galvin L, Inoue Y, Jenkins RG, Johkoh T, Kazerooni EA, Kitaichi M, Knight SL, Mansour G, Nicholson AG, Pipavath SNJ, Buendía-Roldán I, Selman M, Travis WD, Walsh S, Wilson KC; American Thoracic Society; European Respiratory Society; Japanese Respiratory Society; Latin American Thoracic Society. Diagnosis of idiopathic pulmonary fibrosis: an official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med 2018;198:e44-e68.
24. Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, Cottin V, Flaherty KR, Hansell DM, Inoue Y, Kim
DS, Kolb M, Nicholson AG, Noble PW, Selman M, Taniguchi H, Brun M, Le Maulf F, Girard M, Stowasser S, Schlenker-Herceg R, Disse B, Collard HR; INPULSIS Trial Investigators. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 2014;370:2071-2082.
25. King TE Jr, Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, Glassberg MK, Gorina E, Hopkins PM, Kardatzke D, Lancaster L, Lederer DJ, Nathan SD, Pereira CA, Sahn SA, Sussman R, Swigris JJ, Noble PW; ASCEND Study Group. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 2014;370:2083-2092. 26. Ley B, Swigris J, Day BM, Stauffer JL, Raimundo K, Chou
W, Collard HR. Pirfenidone reduces respiratory-related hos-pitalizations in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2017;196:756-761.
27. Costabel U, Inoue Y, Richeldi L, Collard HR, Tschoepe I, Stowasser S, Azuma A. Efficacy of nintedanib in idiopathic pulmonary fibrosis across prespecified subgroups in INPULSIS. Am J Respir Crit Care Med 2016;193:178-185. 28. Nathan SD, Albera C, Bradford WZ, Costabel U, Glaspole I,
Glassberg MK, Kardatzke DR, Daigl M, Kirchgaessler KU, Lancaster LH, Lederer DJ, Pereira CA, Swigris JJ, Valeyre D, Noble PW. Effect of pirfenidone on mortality: pooled analyses and meta-analyses of clinical trials in idiopathic pulmonary fibrosis. Lancet Respir Med 2017;5:33-41.
29. Torrisi SE, Pavone M, Vancheri A, Vancheri C. When to start and when to stop antifibrotic therapies. Eur Respir Rev 2017;26:170053.
30. Collard HR, Ryerson CJ, Corte TJ, Jenkins G, Kondoh Y, Lederer DJ, Lee JS, Maher TM, Wells AU, Antoniou KM, Behr J, Brown KK, Cottin V, Flaherty KR, Fukuoka J, Hansell DM, Johkoh T, Kaminski N, Kim DS, Kolb M, Lynch DA, Myers JL, Raghu G, Richeldi L, Taniguchi H, Martinez FJ. Acute exacerbation of idiopathic pulmonary fibrosis. An inter-national working group report. Am J Respir Crit Care Med 2016;194:265-275.
31. Atkins CP, Loke YK, Wilson AM. Outcomes in idiopathic pulmonary fibrosis: a meta-analysis from placebo controlled trials. Respir Med 2014;108:376-387.
32. Song JW, Hong SB, Lim CM, Koh Y, Kim DS. Acute exacerbation of idiopathic pulmonary fibrosis: incidence, risk factors and outcome. Eur Respir J 2011;37:356-363.
33. Collard HR, Yow E, Richeldi L, Anstrom KJ, Glazer C; IPFnet investigators. Suspected acute exacerbation of idiopathic pulmonary fibrosis as an outcome measure in clinical trials. Respir Res 2013;14:73.
34. Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, Colby TV, Cordier JF, Flaherty KR, Lasky JA, Lynch DA, Ryu JH, Swigris JJ, Wells AU, Ancochea J, Bouros D, Carvalho C, Costabel U, Ebina M, Hansell DM, Johkoh T, Kim DS, King TE Jr, Kondoh Y, Myers J, Müller NL, Nicholson AG, Richeldi L, Selman M, Dudden RF, Griss BS, Protzko SL, Schünemann HJ; ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis. An official ATS/ERS/JRS/
ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011;183:788-824.
35. Ley B, Collard HR, King TE Jr. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2011;183:431-440.
36. King TE Jr, Pardo A, Selman M. Idiopathic pulmonary fibro-sis. Lancet 2011;378:1949-1961.
37. du Bois RM, Nathan SD, Richeldi L, Schwarz MI, Noble PW. Idiopathic pulmonary fibrosis: lung function is a clinically meaningful endpoint for phase III trials. Am J Respir Crit Care Med 2012;186:712-715.
38. Flaherty KR, Mumford JA, Murray S, Kazerooni EA, Gross BH, Colby TV, Travis WD, Flint A, Toews GB, Lynch JP 3rd, Martinez FJ. Prognostic implications of physiologic and radiographic changes in idiopathic interstitial pneumonia. Am J Respir Crit Care Med 2003;168:543-548.
39. Ballester B, Milara J, Cortijo J. idiopathic pulmonary fibrosis and lung cancer: mechanisms and molecular targets. Int J Mol Sci 2019;20:593.
Peer Reviewers’ Commentary
이 논문은 폐의 간질(사이질)에 발생하는 질환을 총칭하는 간질 성 폐질환의 진단 및 치료에 대해서, 가장 흔하고 기대수명이 짧은 특발성 폐섬유증을 중심으로 최신 지식을 정리 설명해 주 고 있다. 광범위한 질환을 포함하는 간질성 폐질환을 임상적으 로 중요한 질환 위주로 분류하고, 그 분류에 따른 진단적 접근 법 및 질환군별 치료법에 대해서도 쉽게 설명하고 있다. 특발성 폐섬유증은 발병기전과 관련된 최신 연구 결과를 소개하여 질 병의 이해도를 높여주고 있으며, 질환의 진행을 지연시키는 효 과가 입증되어 승인된 치료제의 처방 실제 및 부작용 관리까지 제시해 주고 있다. 흉부 CT 검사가 널리 이용되어 간질성 폐질 환의 진단율이 증가하고 있는 상황에서, 이 논문은 어렵고 복잡 한 질환으로만 인식되어 진단 및 치료를 어려움을 겪는 임상 의 사들에게 좋은 정보를 제공해 줄 것으로 판단된다. [정리: 편집위원회]