Crystal Structure of an Activity-enhancing Mutant of DUSP19
Da Gyung Ju, Tae Jin Jeon and Seong Eon Ryu*
Department of Bioengineering, College of Engineering, Hanyang University, Seoul 04763, Korea Received August 14, 2018 /Revised October 5, 2018 /Accepted October 5, 2018
Dual-specificity phosphatases (DUSPs) play a role in cell growth and differentiation by modulating mitogen-activated protein kinases. DUSPs are considered targets for drugs against cancers, diabetes, immune diseases, and neuronal diseases. Part of the DUSP family, DUSP19 modulates c-Jun N-termi- nal kinase activity and is involved in osteoarthritis pathogenesis. Here, we report screening of cav- ity-creating mutants and the crystal structure of a cavity-creating L75A mutant of DUSP19 which has significantly enhanced enzyme activity in comparison to the wild-type protein. The crystal structure reveals a well-formed cavity due to the absent Leu75 side chain and a rotation of the active site- bound sulfate ion. Despite the cavity creation, residues surrounding the cavity did not rearrange significantly. Instead, a tightened hydrophobic interaction by a remote tryptophan residue was ob- served, indicating that the protein folding of the L75A mutant is stabilized by global folding energy minimization, not by local rearrangements in the cavity region. Conformation of the rotated active site sulfate ion resembles that of the phosphor-tyrosine substrate, indicating that cavity creation in- duces an optimal active site conformation. The activity enhancement by an internal cavity and its structural information provide insight on allosteric modulation of DUSP19 activity and development of therapeutics.
Key words : Activity enhancement, allosteric modulation, DUSP19, protein tyrosine phosphatase
*Corresponding author
*Tel : +82-2-2220-4020, Fax : +82-2-2220-4023
*E-mail : [email protected]
This is an Open-Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Life Science 2018 Vol. 28. No. 10. 1140~1146 DOI : https://doi.org/10.5352/JLS.2018.28.10.1140
Introduction
Dual specificity phosphatases (DUSPs) which belong to the protein tyrosine phosphatases (PTPs) family, are classi- fied into six subgroups based on sequence similarity: PRLs (phosphatases of regenerating liver), CDC (Cell division cy- cle) 14 phosphatases, PTENs (phosphatase and tensin homo- logs deleted on chromosome 10), myotubularin, MKPs (mitogen-activated protein kinase phosphatases) and atyp- ical DUSPs [14]. Due to their functions in critical cellular processes, PTPs and DUSPs are considered as drug target against cancers, diabetes, immune diseases, and neuronal diseases [2, 9, 14, 15]. Representative drug development ef- forts by utilizing PTPs/DUSPs include those with PTP1B [10], SHP2 [4] and LMPTP [17] for cancers and diabetes.
DUSP1 and DUSP3 and other DUSPs were also validated as drug targets for depression and immune response impair- ment [6, 11].
DUSP19, which is also called stress-activated protein kin- ase (SAPK) pathway-regulating phosphatase 1 (SKRP1), be- longs to the atypical DUSP and is widely distributed in hu- man tissues [25]. DUSP19 plays a scaffold role for the c-Jun N-terminal kinase (JNK) signaling pathway [25, 26]. Recent studies reported that DUSP19 expression was significantly reduced in osteoarthritis (OA) cartilage and artificial over- expression of DUSP19 restored normal function of chon- drocytes [18]. DUSP19 appeared to inhibit apoptosis of chon- drocytes through dephosphorylating JNK [18]. DUSP19 up-regulation also was shown to inactivate JAK/STAT3 pathway, resulting in inhibition of IL-1β-induced chon- drocytes apoptosis and MMPs expression [24]. Thus, modu- lation of enzyme activity and protein expression of DUSP19 has potential for therapeutics development for OA treatment.
Although PTPs/DUSPs are important therapeutic targets, small molecule drug development efforts targeting the phos- phatase active site have not been easy due to the pocket’s shallow depth and amino acid sequence conservation [16, 20]. Recently, allosteric regulation sites were exploited to overcome difficulties of the active site-targeting approaches [4, 17]. The active site of PTPs/DUSPs appears to be flexible and prone to allosteric regulation [20, 21]. In an effort to understand the effects of allosteric mutations to the enzyme activity, we created various mutations in the hydrophobic
core of the catalytic domain of DUSP19 and analyzed their effects on enzyme activity. Hydrophobic core mutations, which often create internal cavities, are expected to disturb the protein’s structure that may affect enzyme activity allosterically. In case of activity-decreasing mutations, the decreased enzyme activity of hydrophobic core mutations (cavity-creating mutations) can be restored by chemicals [23].
We have previously determined the crystal structure of DUSP19 at high resolutions [19]. The DUSP19 crystals are of the highest quality among structurally-characterized PTPs [19]. Therefore, we exploited DUSP19 to analyze the atom- ic-level effects of hydrophobic core mutations by determin- ing high resolution structures of various activity and struc- ture-modulating mutants. In order to produce cavity in DUSP19, site-directed mutagenesis was performed in the hy- drophobic residues (I69A, L73A, L75A, I95A, L96A and I160A). Most of the cavity-creating mutations exhibited ac- tivity decrease as we expected. However, one mutant (L75A) showed a significant activity increase of 4.2-folds as com- pared to the activity of the wild type enzyme. Leu75 is dis- tant from the active site pocket and the effect to enzyme activity appears to be allosteric. To analyze the structural mechanism of the activity increase, we determined the crys- tal structure of the L75A DUSP19 mutant at 2.29 Å. The structure reveals subtle disturbances in the cavity region that appears to induce an optimal conformation of the active site pocket for phosphorylated substrate-binding. The structure also indicates that the protein folding of the L75A mutant is stabilized by a tightening hydrophobic interaction in a re- mote region. The structural information provides an im- portant example of enzyme activity modulation by structural modification in locations distant from the catalytically active site.
Materials and Methods
Expression and purification
Site directed mutageneses were performed in the vector containing the DUSP19 catalytic domain (residues 65-206, C150S) that was used in the crystallization of the wild type protein [19]. The Stratagene mutagenesis kit was used and mutations were verified by sequencing. The recombinant vector containing the mutated DUSP19 was used to trans- form an E.coli strain BL21 (DE3). Initial expression tests were performed with varying isopropyl β-D-1-thiogalactopyrano-
side (IPTG) concentrations and induction temperatures. The optimal induction condition for overexpression of the L75A mutant was 0.1 mM isopropyl β-D-1-thiogalactopyranoside at 18℃ for overnight. Cells were harvested and resuspended in cell lysis buffer containing 50 mM Tris-Cl (pH7.5), 500 mM NaCl, 1 mM phenylmethylsulfonyl fluoride (PMSF), 0.05%(v/v) 2-mercaptoethanol and 5%(v/v) glycerol and then sonicated. The His-tagged protein was first purified by an affinity chromatography (TALON) column equilibrated with a buffer containing 50 mM Tris-Cl (pH7.5), 500 mM NaCl and 0.5 mM PMSF. After a wash with the equilibration buffer, the bound protein was eluted with a buffer contain- ing the equilibration buffer plus 10 mM β-mercaptoethanol and 0.5 M imidazole. The eluted fractions were dialyzed against a buffer containing 20 mM Tris-Cl (pH 7.5), 50 mM NaCl, 10 mM β-mercaptoethanol and 0.5 mM EDTA. His-tag was removed by thrombin cleavage. The His-tag-removed sample was further purified by Q-Sepharose ion exchange and Sephacryl S-100 gel filtration chromatographies. The fi- nal gel filtration chromatography was performed with a buf- fer containing 25 mM Tris-Cl (pH7.5), 200 mM NaCl, and 2 mM dithiothreitol (DTT). Purified protein was con- centrated to 20 mg/ml, frozen in liquid nitrogen and stored at 203 K.
PTP enzyme assay
The PTP enzyme activity assay was performed using 6,8-difluoro-4-methylumbelliferyl phosphate (DiFMUP) as a substrate. The assay buffer contained 20 mM Tris-Cl (pH 8.0), 0.01% Triton X-100 and 5 mM DTT. Enzyme and sub- strate concentrations were optimized to obtain linear pro- gression curves. The optimized enzyme concentration (same for the wild type and L75A mutant proteins) and substrate (DiFMUP) concentration were 20 μM and 5 μM, respectively.
Fluorescence emission was measured using the fluorescence spectrophotometry (Perkin Elmer, USA) with excitation and emission wavelengths of 335 nm and 460 nm, respectively.
Kinetic parameters were estimated by observing initial ve- locity data and fitting those into the Michaelis-Menten equa- tion using the program SigmaPlot 12.0. All measurements were performed in triplicate.
Crystallization and structure determination
Crystallization was performed at 291 K by using the sit- ting-drop vapor-diffusion method. Initial trials were made under the same conditions as the wild type DUSP19 contain-
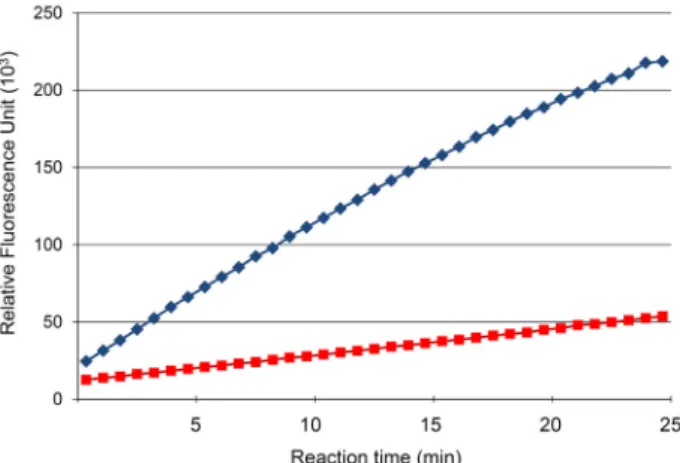
Fig. 1. Progression curves for PTP enzyme reaction. Progression curves of the L75A mutant (blue diamonds, the curve above) and the wild type (red rectangles, the curve be- low) proteins. Concentration of both enzymes were the same (20 mM). X-axis is the reaction time in minutes and Y-axis is the relative fluorescence unit.
Table 1. Data collection and refinement statistics Data collection
Space group Resolution (Å) Cell parameter (Å)
Completeness (%) Rmerge (%) I/σ(I)
P212121
36.09 - 2.29
a=40.31Å, b=47.31Å, c=50.59Å, α=90.0˚, β=90.0˚, γ=90.0˚
95.8 7.8 19.5 Refinement
Number of reflections Number of atoms (protein/nonprotein) Rwork/Rfree (%)
32,070 1,045 / 90
22.86 / 29.71 Rms deviations
Bond distances (Å) Bond angles (˚)
0.0071 0.814 Ramachandran plot Most favored region (%)
Disallowed region (%)
94.74 0.00
ing the active site cysteine mutation (C150S) [19]. Crystals were grown by mixing 1.0 μl of protein (20 mg/ml) solution and an equal volume of reservoir solution containing 1.9-2.0 M ammonium sulfate. Crystals were frozen under nitrogen stream in a cryo-protective buffer containing 2.0 M ammo- nium sulfate and 33% glycerol. The DUSP19-L75A mutant diffracted to 2.29 Å resolution. Diffraction data were col- lected in Pohang Light Source beamline 7A (SBⅠ) [13] and processed by using the HKL 2000 software. The DUSP19- L75A crystal belonged to the P212121 space group as that of the wild type crystal. Unit cell parameters were a=40.31 Å, b=47.31 Å, c=50.59 Å, a=b=g=90.00o. The structure of DUSP- L75A mutant was determined by the molecular replacement technique using the wild type structure as the search target.
The initial electron density map obtained by the program CCP4 [22] confirmed that Leu75 was mutated to alanine. The structure was further refined by the program PHENIX [1]
with rebuildings by using the program COOT [7]. The final refinement statistics are shown in Table 1.
Results and Discussion
Enzyme activity of the L75A mutant
The structure of the wild type DUSP19 [19] was examined and seven hydrophobic core residues with large side chains (Ile69, Leu73, Leu75, Ile95, Leu96 and Ile169) were identified for mutation to alanine. Among the six individual mutant
constructs, only two (I69A and L75A) were found to exhibit high-level expression of soluble proteins. The I69A mutant showed the highest protein expression in the initial ex- pression test. However, large amounts of the protein ag- gregated during the dialysis process with the His-tag affinity purified sample. Although the L75A mutant yielded initial expression lower than the I69A mutant, the mutant did not have the difficulty associated with aggregation, resulting in protein sample sufficient for crystallization.
For the PTP activity assay of the mutants, we screened the optimal reaction condition that yields a linear pro- gression curve (Fig. 1). In the chosen optimal reaction con- dition, the enzyme activity of the L75A mutant was about 4.2-fold higher compared to that of the wild type. Other mu- tant proteins either yielded low protein expression or de- creased activity (data not shown). Because cavity-creation in the protein interior usually accompanies with cavity-filling rearrangements in the cavity area that often propagate to the active site residues, a cavity-creating mutant is expected to result in decrease of enzyme activity. Thus, the activity increase observed in the L75A mutant is unusual and we set out to analyze the crystal structure of the mutant.
Structure of DUSP19-L75A
The L75A mutant (plus the C150S mutation for crystal- lization [19]) was crystallized with the similar crystallization condition to the wild-type protein (see Methods). The C150S mutation was necessary to minimize uncontrolled oxidation of the reactive cysteine as in the crystallization of the wild
A B
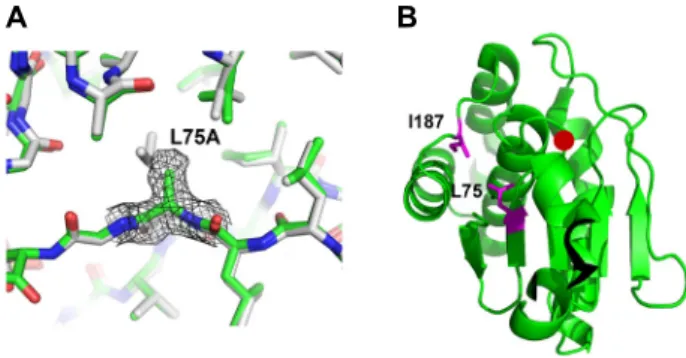
Fig. 2. Conformation and location of the mutated region. (A) The L75A mutant structure (green) is superposed with the wild type structure (grey). The Fo-Fc omit map of residue 75 is presented for the L75A mutant structure.
(B) Locations of the Leu75 (L75) and Ile187 (I187) are indicated as a side chain representation (magenta) on a ribbon diagram of the wild type structure. Position of the active site cysteine is indicated with a red circle.
A B
Fig. 3. Comparison of the active site-bound sulfate ion con- formation. (A) The active site pockets of the L75A mu- tant (green) and the wild type (magenta) structures are superposed. The sulfate ion (SO42-
) of the L75A mutant structure is represented in cyan. (B) The active site pock- et structure of the phosphorylated substrate-DUSP3 complex (blue) is superposed with that of the L75A mu- tant (green). As in (a), The sulfate ion of the L75A mu- tant structure is represented in cyan. To show the phos- phor-tyrosine (P-Tyr) better, the active site pocket re- gion was slightly rotated compared to (a).
type [19]. The structure of the L75A mutant was determined by the molecular replacement technique and refined to a res- olution of 2.29 Å (Table 1). In the electron density map, the cavity due to the absence of the Leu75 side chain was evident. Unlike the previously-reported I187A mutant struc- ture, which also was designed to create an internal cavity [8], there was no major cavity-filling rearrangement of main chain atoms. In the structure of the I187A mutant, the I187A-containing loop moved significantly towards the cav- ity to reduce the size of cavity [8]. In comparison, the cav- ity-facing residues of the L75A structure did not show sig- nificant movements. When the L75A mutant structure was superposed with the wild type structure, the displacement between the two structures was negligible (Fig. 2). This may be due to the different interactions of the two mutated resi- dues with neighboring main chains (Fig. 2): I187A is in the loop between two helices, and there are little main chain hydrogen bonds restricting the loop conformation. In com- parison, L75A is located in a β-strand that is a part of the central b-sheet and the main chain of the L75A-containing strand makes numerous hydrogen bonds with neighboring β-strands for the β-sheet formation. Thus, it would be diffi- cult for the L75A containing region to move towards the cavity.
Due to the lack of main chain movements in the L75A mutant, the cavity size is significantly larger than that of the I187A mutant. In the L75A mutant, there was a rotation in the side chain of Val161 that placed a C-gamma atom of the residue a little towards the cavity (Fig. 2). However, the cavity-filling effect by the Val161 rotation is minimal.
The cavity formed by the L75A mutation faces the catalytic P-loop and results in subtle changes in the loop. The side chain of Val154 is rotated by 180 degrees affecting the active pocket-bound sulfate ion (Fig. 3). In comparison to the wild type structure, the active site sulfate ion of the L75A mutant is a little displaced towards the pocket entrance and the ori- entation of oxygen atoms are reversed. These differences in the sulfate ion appears due to the rotation of Val154.
Superposition of the L75A structure with the phosphory- lated substrate-bound DUSP3 structure revealed that the sul- fate ion orientation matches with that of phosphate moiety in the phosphorylated substrate, indicating that the active site of the L75A is better suited for dephosphorylation reaction. The similar conformation changes in Val154 and sulfate ion were observed in the I187A mutant, too [8], sug- gesting that the Val154 conformation is critical to align the phosphate group of the phosphorylated protein substrates.
However, in the I187A structure, the main chain movement caused by the cavity rearrangement appears to induce other differences in the P-loop residues that may result in decrease of enzyme activity of the mutant [8].
Stabilization of a remote region
Although conformation changes of residues surrounding the cavity are minimal and subtle in the L75A mutant, sig- nificant conformation changes were observed in a region re- mote from the cavity. A careful examination of superposed
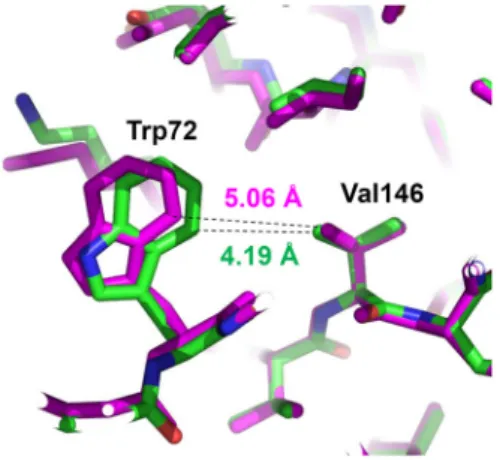
Fig. 4. Movement of a remote tryptophan. The region of the tryptophan movement is shown with superposition be- tween structures of the L75A mutant (green) and the wild type (magenta). Interatomic distances between closest atoms of Trp72 and Val146 are indicated for the two structures.
structures of the L75A mutant and the wild type revealed that the side chain of Trp72 moved significantly towards the hydrophobic core of the protein (Fig. 4). The Trp72 move- ment did not occur in the structure of the I187A mutant (PDB code: 4D3P) [8]. In the wild type structure, the Trp72 side chain is loosely packed onto the hydrophobic core con- sisting of Val146, Leu173 and Ile136. The closest distance be- tween the neighboring side chains of Trp72 and Val146 is 5.06 Å in the wild type, whereas the distance is shortened to 4.19 Å, resulting in a tighter hydrophobic interaction in the L75A mutant. The distance between Trp72 and Ile136 side chains is also shortened (Fig. 4).
It is striking to observe a large movement of Trp72 that is remote from the cavity when the cavity-facing hydro- phobic residues including Val146, Leu173 and Ile136 did not move. In general, allosteric switches in protein structures oc- cur through sequential changes in the intervening residues [8]. In the case of Trp72 movement in the L75A mutant, there was no movement or relay of the intervening residues. Thus, the Trp72 movement is likely due to a global energy mini- mization rather than an interaction energy-driven transition.
Because the internal cavity in proteins destabilizes protein structure [3], the tighter interaction between hydrophobic residues due to the Trp72 movement may offset the destabi- lization by the L75A cavity formation. The energy compen- sation by stabilization of a remote region seen in the L75A structure indicates that proteins function as a global entity where many parts of the protein communicate with each other by a global energy balance. It will be interesting to
analyze the folding process and dynamics of the L75A mu- tant in future studies.
Cavity-induced activity modulation
Creation of a cavity in proteins triggers conformational changes of cavity-proximal residues to fill the internal va- cancy [8]. The movement of cavity-proximal residues results in another vacancy in the opposite side that induces other movements of next residues. These successive movements can eventually affect conformation of residues participating in enzyme reaction. For example, the previously analyzed cavity-creation mutant of DUSP19 (I187A) revealed cavity- filling rearrangements involving movement of the mutated Ala187-containing loop towards the cavity and consecutive movement of Val154 and other residues in the P-loop. As a result of those movement, the PTP enzyme activity of the DUSP19-I187A mutant decreased by about 40%[8].
The 4.2-fold activity increase observed in the case of the L75A mutant is different from what one usually would expect. There may be two possible explanations for the activ- ity increase. The first is the conformational changes of Val154 in the P-loop resulting in the shift of the sulfate bind- ing geometry towards a more suited one for catalysis as.
The effect of the shift of the sulfate ion binding geometry appears complex because the activity-decreasing I187A mu- tant also showed the similar geometry of sulfate ion [8]. In the case of the I187A mutant, however, large cavity-filling rearrangements are likely to induce subtle differences in con- formation and dynamics of the P-loop, which could cause decrease the enzyme activity.
The next possible explanation for the increased activity may be the induced dynamics of the enzyme due to the in- ternal cavity. Protein flexibility is required for optimal activ- ity of enzymes [12]. The enzyme catalysis by protein tyrosine phosphatase 1B (PTP1B) also involves structural dynamics of several regions of the protein [5]. Thus, the cavity-induced activity increase of the L75A mutant of DUSP19 may be caused by the cavity-induced dynamics of the P-loop regions. In the case of the activity-decreasing I187A mutant, the cavity volume was decreased by the cavity-filling re- arrangements to lessen the cavity-induced dynamics [8].
Thus, the activity increase of the L75A mutant appears to be originated from induction of the optimal active site con- formation as well as the cavity-induced dynamics in the ac- tive site region. The L75A mutant had a larger fold-differ- ence in Kcat increase (2.7-fold) than KM decrease (2.3-fold)
Table 2. Enzyme kinetics
Vmax (RFU min-1) KM (mM-1) Kcat (RFU min-1 mM-1) Kcat/KM
Wild type L75A
8,477±373 23,521±822
51.9±6.0 22.8±2.6
423 1,176
8.1 51.5
(Table 2), indicating that the activity enhancement by con- formational flexibility was more influential than that by changes in active site structure. Thus, kinetics observations are consistent with the results obtained from structural analysis. However, the definite conclusion on the con- tribution of cavity-induced dynamics would require further biochemical and computational analyses. Because DUSP19 was shown to have potential as a protein therapeutic for OA treatment [18], the activity-increasing L75A mutant can be developed as an efficient therapeutic protein and the structural information of the mutant can contribute to fur- ther designing of activity modulating variants of DUSP19 and related proteins.
References
1. Adams, P. D., Afonine, P. V., Bunkoczi, G., Chen, V. B., Davis, I. W., Echols, N., Headd, J. J., Hung, L. W., Kapral, G. J., Grosse-Kunstleve, R. W., McCoy, A. J., Moriarty, N.
W., Oeffner, R., Read, R. J., Richardson, D. C., Richardson, J. S., Terwilliger, T. C. and Zwart, P. H. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213-221.
2. Bermudez, O., Pages, G. and Gimond, C. 2010. The du- al-specificity MAP kinase phosphatases: critical roles in de- velopment and cancer. Am. J. Physiol. Cell Physiol. 299, C189- 202.
3. Boopathy, S., Silvas, T. V., Tischbein, M., Jansen, S., Shandilya, S. M., Zitzewitz, J. A., Landers, J. E., Goode, B.
L., Schiffer, C. A. and Bosco, D. A. 2015. Structural basis for mutation-induced destabilization of profilin 1 in ALS.
Proc. Natl. Acad. Sci. USA. 112, 7984-7989.
4. Chen, Y. N., LaMarche, M. J., Chan, H. M., Fekkes, P., Garcia-Fortanet, J., Acker, M. G., Antonakos, B., Chen, C.
H., Chen, Z., Cooke, V. G., Dobson, J. R., Deng, Z., Fei, F., Firestone, B., Fodor, M., Fridrich, C., Gao, H., Grunenfelder, D., Hao, H. X., Jacob, J., Ho, S., Hsiao, K., Kang, Z. B., Karki, R., Kato, M., Larrow, J., La Bonte, L. R., Lenoir, F., Liu, G., Liu, S., Majumdar, D., Meyer, M. J., Palermo, M., Perez, L., Pu, M., Price, E., Quinn, C., Shakya, S., Shultz, M. D., Slisz, J., Venkatesan, K., Wang, P., Warmuth, M., Williams, S., Yang, G., Yuan, J., Zhang, J. H., Zhu, P., Ramsey, T., Keen, N. J., Sellers, W. R., Stams, T. and Fortin, P. D. 2016.
Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature 535, 148-152.
5. Choy, M. S., Li, Y., Machado, L., Kunze, M. B. A., Connors,
C. R., Wei, X., Lindorff-Larsen, K., Page, R. and Peti, W.
2017. Conformational rigidity and protein dynamics at dis- tinct timescales regulate PTP1B activity and allostery. Mol.
Cell 65, 644-658 e645.
6. Duric, V., Banasr, M., Licznerski, P., Schmidt, H. D., Stock- meier, C. A., Simen, A. A., Newton, S. S. and Duman, R.
S. 2010. A negative regulator of MAP kinase causes depres- sive behavior. Nat. Med. 16, 1328-1332.
7. Emsley, P., Lohkamp, B., Scott, W. G. and Cowtan, K. 2010.
Features and development of Coot. Acta Crystallogr. D Biol.
Crystallogr. 66, 486-501.
8. Jeon, T. J., Nam, K. T. and Ryu, S. E. 2015. Structural analy- sis of activity-modulating mutations of DUSP19. Biodesign 3, 111-116.
9. Keyse, S. M. 2008. Dual-specificity MAP kinase phospha- tases (MKPs) and cancer. Cancer Metastasis Rev. 27, 253-261.
10. Krishnan, N., Koveal, D., Miller, D. H., Xue, B., Akshinthala, S. D., Kragelj, J., Jensen, M. R., Gauss, C. M., Page, R., Blackledge, M., Muthuswamy, S. K., Peti, W. and Tonks, N. K. 2014. Targeting the disordered C terminus of PTP1B with an allosteric inhibitor. Nat. Chem. Biol. 10, 558-566.
11. Li, G., Yu, M., Lee, W. W., Tsang, M., Krishnan, E., Weyand, C. M. and Goronzy, J. J. 2012. Decline in miR-181a ex- pression with age impairs T cell receptor sensitivity by in- creasing DUSP6 activity. Nat. Med. 18, 1518-1524.
12. Mukherjee, J. and Gupta, M. N. 2015. Increasing importance of protein flexibility in designing biocatalytic processes.
Biotechnol. Rep. (Amst) 6, 119-123.
13. Park, S. Y., Ha, S. C. and Kim, Y. G. 2017. The protein crys- tallography beamlines at the Pohang Light Source II.
Biodesign 5, 30-34.
14. Patterson, K. I., Brummer, T., O'Brien, P. M. and Daly, R.
J. 2009. Dual-specificity phosphatases: critical regulators with diverse cellular targets. Biochem. J. 418, 475-489.
15. Pulido, R. and Hooft van Huijsduijnen, R. 2008. Protein ty- rosine phosphatases: dual-specificity phosphatases in health and disease. FEBS J. 275, 848-866.
16. Ryu, S. E. and Kim, S. J. 2014. Targeting allosteric sites for protein tyrosine phosphatase inhibition. Biodesign 2, 81-90.
17. Stanford, S. M., Aleshin, A. E., Zhang, V., Ardecky, R. J., Hedrick, M. P., Zou, J., Ganji, S. R., Bliss, M. R., Yamamoto, F., Bobkov, A. A., Kiselar, J., Liu, Y., Cadwell, G. W., Khare, S., Yu, J., Barquilla, A., Chung, T. D. Y., Mustelin, T., Schenk, S., Bankston, L. A., Liddington, R. C., Pinkerton, A. B. and Bottini, N. 2017. Diabetes reversal by inhibition of the low-molecular-weight tyrosine phosphatase. Nat. Chem. Biol.
13, 624-632.
18. Wang, Y., Xu, Z., Wang, J. and Xu, S. 2016. DUSP19, a down- stream effector of leptin, inhibits chondrocyte apoptosis via dephosphorylating JNK during osteoarthritis pathogenesis.
초록:효소활성 증가 돌연변이를 함유한 DUSP19의 결정구조
주다경․전태진․류성언*
(한양대학교 공과대학 생명공학과)
이중탈인산화효소(DUSP)는 성장인자활성 단백질키나제(MAPK)를 조절해서 세포성장과 분화에 관여하며 암, 당뇨병, 면역질환, 신경질환의 신약개발표적이다. DUSP 단백질군에 속하는 DUSP19는c-Jun N-말단 키나제(JNK) 를 조절하며 골관절염의 질환화과정에 관여한다. 우리는 야생형 DUSP19 에 비하여 상당히 활성이 증가된 cavity 형성 돌연변이인 DUSP19-L75A의 결정구조를 규명하였다. 결정구조는 Leu75의 곁가지가 없어진 결과로 cavity 가 잘 형성되어 있는 것을 보여주며, 활성부위에 결합한 황이온이 회전된 형태로 존재하는 것을 보여준다. Cavity 형성에도 불구하고 cavity를 둘러싸고 있는 잔기들은 그다지 재조정되지 않은 것으로 나타나며 그 대신에 멀리 떨어진 트립토판 잔기가 소수성결합을 강화하고 있는 것으로 나타나서 L75A 돌연변이의 접힘은 cavity 부위의 재조정이 아니라 글로벌 접힘 에너지 최소화 기작에 의해 안정화 되었음을 발견할 수 있었다. 회전된 활성화부위 황이온의 구조는 인산화티로신 잔기와 유사함이 발견되어 L75A 돌연변이가 최적의 활성화형태를 유도했다는 것 을 알 수 있었다. 내부 cavity에 의한 활성증가현상과 이에 대한 구조적 정보는 DUSP19의 알로스테릭 조절과 치 료제 개발에 정보를 제공한다.
Mol. Biosyst. 12, 721-728.
19. Wei, C. H., Ryu, S. Y., Jeon, Y. H., Yoon, M. Y., Jeong, D.
G., Kim, S. J. and Ryu, S. E. 2011. Crystal structure of a novel mitogen-activated protein kinase phosphatase, SKRP1.
Proteins 79, 3242-3246.
20. Wei, C. H. and Ryu, S. E. 2016. Structural mobility of the active site in classical protein tyrosine phosphatases and du- al specificity phosphatases. Biodesign 4, 77-87.
21. Wei, C. H., Min, H. G., Kim, M., Kim, G. H., Chun, H. J.
and Ryu, S. E. 2018. Two intermediate states of the con- formational switch in dual specificity phosphatase 13a.
Pharmacol. Res. 128, 211-219.
22. Winn, M. D., Ballard, C. C., Cowtan, K. D., Dodson, E. J., Emsley, P., Evans, P. R., Keegan, R. M., Krissinel, E. B., Leslie, A. G. W., McCoy, A., McNicholas, S. J., Murshudov, G. N., Pannu, N. S., Potterton, E. A., Powell, H. R., Read, R. J., Vagin, A. and Wilson, K. S. 2011. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235-242.
23. Xia, Y., DiPrimio, N., Keppel, T. R., Vo, B., Fraser, K.,
Battaile, K. P., Egan, C., Bystroff, C., Lovell, S., Weis, D.
D., Anderson, J. C. and Karanicolas, J. 2013. The desig- nability of protein switches by chemical rescue of structure:
mechanisms of inactivation and reactivation. J. Am. Chem.
Soc. 135, 18840-18849.
24. Yao, Z. Z., Hu, A. X. and Liu, X. S. 2017. DUSP19 regulates IL-1beta-induced apoptosis and MMPs expression in rat chondrocytes through JAK2/STAT3 signaling pathway.
Biomed. Pharmacother. 96, 1209-1215.
25. Zama, T., Aoki, R., Kamimoto, T., Inoue, K., Ikeda, Y. and Hagiwara, M. 2002. A novel dual specificity phosphatase SKRP1 interacts with the MAPK kinase MKK7 and in- activates the JNK MAPK pathway. Implication for the pre- cise regulation of the particular MAPK pathway. J. Biol.
Chem. 277, 23909-23918.
26. Zama, T., Aoki, R., Kamimoto, T., Inoue, K., Ikeda, Y. and Hagiwara, M. 2002. Scaffold role of a mitogen-activated pro- tein kinase phosphatase, SKRP1, for the JNK signaling path- way. J. Biol. Chem. 277, 23919-23926.