Treosulfan-Based Conditioning Regimen for Hematopoietic Stem Cell Transplantation in Pediatric Patients with Hemophagocytic Lymphohistiocytosis
Ho Joon Im and Sung Han Kang
Department of Pediatrics, University of Ulsan College of Medicine, Asan Medical Center Children’s Hospital, Seoul, Korea
Hemophagocytic lymphohistiocytosis (HLH) is a fatal disease unless timely and effec- tive treatment is given. Immunochemotherapy including etoposide, with or without hematopoietic stem cell transplantation (HCT), has improved the outcomes for pa- tients with HLH. In patients with familial or refractory HLH, HCT is now routinely performed and is the only curative treatment. Conditioning regimens play an im- portant role in the success of HCT for pediatric patients with HLH and other non- malignant diseases and have improved dramatically in recent decades. The initial HCT approach using myeloablative conditioning significantly improved the outcomes of patients with HLH but was associated with considerable transplant-related mor- tality. A subsequent strategy using reduced-intensity conditioning (RIC) remarkably reduced the incidence of T RM. However, the high level of mixed chimerism asso- ciated with RIC has prompted the search for improved conditioning regimens. Re- cently, treosulfan has replaced busulfan as a component of a reduced toxicity con- ditioning regimen. Both its myeloablative and immunosuppressive properties, as well as a favorable toxicity profile, make treosulfan a potential candidate for use as part of conditioning regimen prior to HCT. Indeed, treosulfan-based conditioning regi- mens are being increasingly used in pediatric patients with various non-malignant diseases. We here review the recent progress in HCT for pediatric HLH with a focus on treosulfan-based conditioning regimens, including our own experience.
pISSN 2233-5250 / eISSN 2233-4580 https://doi.org/10.15264/cpho.2021.28.1.28 Clin Pediatr Hematol Oncol 2021;28:28∼38
Received on March 29, 2021 Revised on April 22, 2021 Accepted on April 22, 2021
Corresponding Author: Ho Joon Im Department of Pediatrics, University of Ulsan College of Medicine, Asan Medical Center, 88-1 Olympic-ro 43-gil, Song pa-g u, Seoul 05505, Korea Tel: +82-2-3010-3371
Fax: +82-2-473-3725 E-mail: [email protected] ORCID ID: orcid.org/0000-0001-8799-4068 Key Words: Hematopoietic cell transplantation, Conditioning regimen, Treosulfan,
Hemophagocytic lymphohistiocytosis, Children and adolescents
Copyright ⓒ 2021 Korean Society of Pediatric Hematology-Oncology
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/
Introduction
Hemophagocytic lymphohistiocytosis (HLH) is a clin- icopathological entity characterized by an impaired or absent function of NK cells and cytotoxic T cells and un- controlled inflammation, as well as being the most prev- alent and clinically significant macrophage-related his- tiocytic disorder [1,2]. Dysregulation in immune system function results in uncontrolled and ineffective immune activation, leading to cellular damage and multiorgan
dysfunction as well as the proliferation and activation of benign macrophages and causing pancytopenia, hep- atosplenomegaly, and lymphadenopathy [3]. HLH com- prises two different conditions, primary or genetic HLH and secondary or acquired HLH, which may be difficult to distinguish from each other [4-6]. Primary HLH is fur- ther divided into familial HLH (FHL1-5: PRF1, UNC13D, STX11, STXBP2), in which HLH is the only manifestation of the disease, and other genetic causes including X-linked lymphoproliferative disease (XLP), Griscelli syn- drome (GS, RAB27A), and Chediak-Higashi syndrome
(CHS, LYST) in which HLH is one of several clinical mani- festations [7].
Immunochemotherapy including etoposide, with or without hematopoietic stem cell transplantation (HCT), has improved the outcomes of patients with HLH, includ- ing FHL [8,9]. In patients with familial or refractory HLH, HCT is now routinely performed and is the only curative treatment [7,10,11]. Conditioning regimens play important roles for successful HCT. Earlier experiences with HCT using conventional myeloablative conditioning (MAC) have shown this approach to be associated with an ex- cessively high rate of toxicity and death [12]. Reduced- intensity conditioning (RIC) was introduced in an attempt to improve the outcome of HCT for HLH. RIC signifi- cantly reduced the rates of transplant-related mortality (TRM) but was found to be associated with a high in- cidence of mixed chimerism requiring subsequent inter- ventions [13,14].
The goal of conditioning in children with non-malig- nant disease is the cure of underlying disease with mini- mal short-term and long-term toxicities. The high TRM with MAC and high mixed chimerism associated with RIC have prompted the search for new conditioning regimens for HCT in the treatment of HLH. Recently, treosulfan has replaced busulfan as a reduced toxicity conditioning (RTC) regimen. Treosulfan is a prodrug of a bifunctional alkylating cytotoxic agent with low organ toxicity in pe- diatric patients with malignant or non-malignant dis- eases [15,16].
We here review the recent progress in HCT for pedia- tric patients with HLH, focusing on treosulfan-based conditioning regimens prior to transplant. We also dis- cuss our own experiences with this approach.
Review of Hematopoietic Cell Transplantation for HLH
Although the introduction of the HLH-94 treatment protocol dramatically improved the outcomes for pa- tients with HLH, HCT is needed in selected cases and is regarded as a curative treatment for patients with re- fractory, or relapsed cases, as well as primary HLH.
HLH-94, which is the first prospective international study of HLH, significantly improved the clinical outcomes, with a reported 3-year overall survival (OS) of 55% for whole cohort and 62% who received HCT [8]. Horne et al. reported their HCT results who were treated with the HLH-94 protocol [12] and indicated that a MAC regimen consisting of busulfan, cyclophosphamide and etoposide produced a 3-year overall survival (OS) of 64%, but TRM rate of 30%, which is higher than described for other non-malignant disease [17-19]. Other subsequent studied reported similar OS ranging from 52% to 75% (Table 1) [20-23]. However, the obvious major limitation of the HCT using MAC was a high TRM ranging from 17% to 35%.
Given the high frequency of TRM with MAC, a reduced intensity conditioning (RIC) regimen was introduced to reduce the TRM. Cooper et al. reported a favorable out- come using RIC with fludarabine and melphalan with or without serotherapy [24]. Nine of 12 patients in that study cohort survived but 3 of these 9 survivors showed mixed chimerism. A report from Cincinnati Children’s Hospital comparing the post-transplant outcomes of pa- tients with HLH in accordance with the conditioning reg- imens concluded that RIC significantly improved the sur- vival, which was attributable to a reduction in early mor- tality after HCT [13]. The conditioning regimens used in the patient populations in that report consisted of flu- darabine, melphalan and alemtuzumab for RIC (n=26), and busulfan, cyclophosphamide, and rabbit ATG (r- ATG, Thymoglobulin) with or without etoposide for MAC (n=14). The estimated 3-year survival was significantly better for the RIC compared to MAC (92% vs. 43%, P=0.001), but mixed chimerism was significantly more frequent in RIC than MAC (65% vs. 18%, P=0.011). Most of those patients showing mixed chimerism received an intervention involving an early weaning of immunosup- pressants given for graft versus host disease (GVHD) pro- phylaxis and/or the use of donor lymphocyte infusion (DLI) to stabilize or increase the level of donor chime- rism. A subsequent study utilizing the same conditioning with fludarabine, melphalan and alemtuzumab reported that only 41% of HLH patients survived on stable engraft-
ment without any interventions [25]. Observations in oth- er cohorts have indicated improved outcomes with RIC over the MAC regimen but found also that the prevalence of mixed chimerism was higher after transplantation when the RIC regimen was used (Table 1) [26,27]. In con- clusion, RIC regimens improved the outcomes of HCT for HLH in terms of a lower TRM but were associated with higher incidence of mixed chimerism requiring DLI or salvage transplantation.
Treosulfan as a Component of Conditioning Regimen
The high TRM associated with MAC, and high mixed chimerism resulting from RIC, have prompted the search for better conditioning regimens for HCT in the treat- ment of HLH. Treosulfan, which is a busulfan analogue, is a prodrug and a water-soluble bifunctional alkylating agent which has been used as treatment for various can- cers [15,28-30]. Recently, treosulfan replaced busulfan as a component of a reduced toxicity conditioning regimen for HCT. Preclinical studies demonstrated that treosulfan treatment produced a rapid and sustained myelosup- pression and that this agent exhibits immunosuppressive characteristics, which contribute to stable engraftment after HCT [15,31-33]. In contrast to busulfan, treosulfan is also associated with fewer extramedullary toxicities, including in the liver. Both its myeloablative and im- munosuppressive properties, as well as its favorable tox- icity profile, make treosulfan a potentially viable compo- nent of a more optimal conditioning regimen prior to HCT.
Treosulfan-Based Conditioning Regimen in Pediatric Patients with Non-Malignant Disease
Treosulfan-based conditioning regimens have increas- ingly been used in pediatric patients with various non- malignant diseases (Table 2) [34-41]. Treosulfan with flu- darabine has been the most frequent combination used as a conditioning regimen for HCT in prior clinical trials.
Various combinations of treosulfan-based conditioning Table 1. Selected reports on allogeneic hematopoietic stem cell transplantation for hemophagocytic lymphohistiocytosis Study/authorYearNMajor conditioning regimenVOD (n or %)Mixed chimerismGVHD TRMSurvivalRef Acute (grade)Chronic Myeloablative conditioning (MAC) HLH-94/Horne200586Bu, Cy, VP4 (dead)19%32% (2-4)9%30%64% (3y)12 Baker200891Bu, Cy, VP±ATG18%11%41% (2-4)25%35%52% (1y), 45% (3y)20 AIEOP/Cesaro200861Bu, Cy±VP7 (severe)15%31% (2-4)17%26%59% (8y)21 Japan/Ohga201043Bu, Cy, VP±ATGNS19%NSNS17%65% (10y)22 Korea/Seo201019Bu, Cy, VP±ATG2NS26% (2-4)NS20%75% (5y)23 CCHMC/Marsh201014Bu, Cy ATG±VP018%14% (2-3)NS57%43% (3y)13 Reduced intensity conditioning (RIC) UK/Cooper200612Flu, Mel, Alem or ATG033%33% (2-4)33%NS75% (2.5y)24 UK/Cooper200825Flu, Mel, Alem or ATG029%NSNSNS84% (3y)26 CCHMC/Marsh201026Flu, Mel, Alem065%8% (2-3)NS12%92% (3y)13 USA/Allen201834Flu, Mel, Alem059%*27% (2-4)NS∼29%82% (1y), 68% (1.5y)25 *Patients with mixed chimerism or received additional interventions. Bu, busulfan 16 mg/kg; Cy, cyclophosphamide 120-200 mg/kg; VP, etoposide 30 mg/kg or 900 mg/m2 ; ATG, antithymocyte globulin; Flu, fludarabine 150 mg/m2 ; Mel, melphalan 140 mg/m2 ; Alem, alemtuzumab; VOD, veno-occlusive disease; TRM, transplant-related mortality; NS, not specified; Ref, reference.
Table 2. Selected reports on hematopoietic stem cell transplantation after treosulfan-based conditioning in pediatric patients with non-malignant disease Study/authorYearNDiagnosis (n)Main conditioning regimensVOD (n or %)Mixed chimerismGVHD (n or %)TRM (n or %)SurvivalRef Acute (gr)Chronic Lehmberg201419HLH (19)Treo, Flu, Alem±TT153%6% (3)0%0%100%45 Burroughs201431PID (13), HLH (6), DBA (3), SDS (3), SCD (4), PNH (2)Treo, Flu±TT0%29% (CD3+ )10% (3-4)21%10%90%36 Dinur-Schejter201544PID (27), hematologic (9), metabolic (9)Treo, Flu±TTNS28%27% (3-4)19%1271%37 EBMT/Slatter2015316Inherited disorders (188), hemoglobinopathy (70), histiocytic disorder (32), BMF (24), others (2) Treo, Flu±TT Treo, Cy0% (≥grade 3)NS10% (3-4)NS5191% for thalassemia, 100% for SCD
38 Morillo- Gutierrez201670CGD (70)Treo, Flu±TT±ATG or Alem0%25% (CD3+ ), 20% (CD15+ )12% (3-4)13%691.4%39 FHCRC/ Burroughs201714SDS (3), DBA (4), PNH (4), others (3)Treo, Flu0%7%1 (4)2113 alive40 Slatter2018160SCID (39), WAS (20), CGD (17), HLH (18), others (66)Treo, Flu, Alem0%Various9% (3-4)242783%41 HLH, hemophagocytic lymphohistiocytosis; PID, primary immunodeficiency; DBA, Diamond-Blackfan anemia; SDS, Shwachman-Diamond syndrome; SCD, sickle cell disease; PNH, paroxysmal nocturnal hemoglobinuria; BMF, bone marrow failure; CGD, chronic granulomatous disease; SCID, severe combined immunodeficiency; WAS, Wiskott-Aldrich syndrome; VOD, veno-occlusive disease; TRM, transplant-related mortality; Treo, treosulfan 36 or 42 g/m2 ; Flu, fludarabine 150-180 mg/m2 ; TT, thiotepa 7-10 mg/kg; ATG, antithymocyte globulin; Alem, alemtuzumab; Cy, cyclophosphamide 200 mg/kg; NS, not specified; Ref, reference.
Fig. 1. Various combinations of treosulfan-based conditioning regi- mens. TREO, treosulfan; FLU, flu- darabine; CY, cyclophosphamide;
VP16, etoposide; MEL, melphalan;
TT, thiotepa; TBI, total body irra- diation.
regimens are shown in Fig. 1. In 2008, HCT using treo- sulfan-containing regimens showed high rates of en- graftment with low RRT and transplant mortality in 32 children with non-malignant disease but with significant comorbidities [34]. Fludarabine and alemtuzumab were added to treosulfan as conditioning regimen in 22 pedia- tric patients in that study and 26 patients received treo- sulfan at a dose of 42 g/m2. Twenty-seven of the patients in the entire cohort survived with an OS of 84%. EBMT data for a large number of pediatric patients with non- malignant disease demonstrated that treosulfan-based conditioning is a safe and effective regimen even for children younger than 1 year at the time of transplan- tation [38]. Among the 316 patients in that study, the total doses of treosulfan were <33 g/m2 in 15, 33-39 g/m2 in 90, and >39 g/m2 in 211 patients. Toxicity was not found to be associated with an increasing dose of treosulfan and the OS was similar among the three dosage groups.
Furthermore, the addition of thiotepa to the treosulfan and fludarabine regimen did not increase acute toxicity levels. A report incorporating thiotepa into a fludar- abine/melphalan/alemtuzumab regimen demonstrated the improved engraftment [42], although another study of conditioning regimen with additional thiotepa showed a beneficial role in chimerism but in the survival [37].
A prospective multicenter trial of treosulfan-based conditioning for 31 patients with various non-malignant diseases demonstrated an excellent outcome without po-
tentially serious complications [36]. Fludarabine±thymo- globulin was combined with treosulfan as part of that conditioning regimen. All patients showed successful en- graftment, a TRM of 0% at day-100, and an OS of 90%.
In 2018, Slatter et al. reported a favorable OS of 87.1%
with a high level of complete or stable mixed chimerism after conditioning with treosulfan and fludarabine, with or without alemtuzumab, for HCT in 160 children with primary immunodeficiency [41]. In that study, the com- bination of treosulfan with fludarabine was utilized based on the authors’ previous report that revealed less toxicity and better chimerism with fludarabine compared to cy- clophosphamide [35]. Severe combined immunodeficiency (SCID, n=39), Wiskott-Aldrich syndrome (WAS, n=20), chronic granulomatous disease (CGD, n=17), and HLH (n=18) were the major diseases among the 160 patients analyzed, and 79% of patients received HCT from un- related donors. Treosulfan was given at a dose of 42 g/m2 (n=102) but was reduced to 36 g/m2 (n=54) for infants aged <1 year. The survivals for the SCID, WAS and CGD were all excellent at above 90%, but the outcome for the HLH was not as satisfactory with an OS of 63%. Around 70% of the patients in that study who received peripheral blood stem cells achieved >95% donor chimerism.
The cumulative evidence for the impacts of treosulfan- based RTC for pediatric patients with non-malignant dis- ease indicates that this conditioning regimen is well tol- erated and has a low toxicity profile. Treosulfan-based
RTC regimens have thus emerged as a more optimal con- ditioning method for HCT in pediatric patients with vari- ous non-malignant diseases. Although treosulfan is wide- ly being used as part of conditioning prior to HCT in children with malignant or non-malignant diseases, the studies on pharmacokinetics (PK) of treosulfan are scarce [43,44]. Personalized dosing of treosulfan is crucial, es- pecially in pediatric patients, to minimize toxicity and avoid rejection or relapse of underlying disease. To date, the most optimal dosing of the drug based on pharmaco- kinetic data remains unclear. Future study on the PK of treosulfan will further improve the outcome of HCT in pediatric patients.
Treosulfan-Based Conditioning Regimen for HLH
The outcomes of HCT using treosulfan-based con- ditioning regimen for HLH have mostly been reported in cohorts that included patients with a variety of non-ma- lignant diseases. A few studies have been published on the results of HCT using this conditioning regimen in HLH patients only.
Lehmberg et al. reported on the HCT using treosulfan- based conditioning in 19 children and adolescents with HLH [45]. Twelve of these patients had familial HLH and 7 had other genetic defects. The conditioning regimen consisted of treosulfan (42 g/m2 or 36 g/m2 if <12 kg), fludarabine (150-180 mg/m2 or 5-6 mg/kg), facultative thiotepa (10 mg/kg or 7 mg/kg if <12 kg), and alem- tuzumab. The toxicity profile was favorable and all en- rolled 19 patients survived. Notably however, 8 (42%) of these patients required additional cellular therapy such as DLI, stem cell boost, or a second HCT, which was a high rate for a cohort with HLA-mismatched donors, in- cluding one patient receiving haploidentical HCT. The inclusion of thiotepa and/or adjustment of the serother- apy was suggested in attempts to improve the primary complete donor chimerism in recipients of a graft from a HLA-mismatched donor. A subsequent report indicated that a HLA-mismatched donor and active disease at the start of conditioning were the only risk factors for sub-
stantial recipient chimerism [46].
There have been a few studies comparing the out- comes of treosulfan-based conditioning with other con- ditioning regimen in HLH. Messina et al. reported the re- sults of HCT for 109 patients with HLH including 31 FHL2 and 32 FHL3 cases [47]. The conditioning regimens in this cohort included a busulfan-based regimen in 61 pa- tients, fludarabine-based in 26 cases, and treosulfan- based in 21 cases. Although no statistically significant differences were observed in that study, a trend toward a better OS and event-free survival (EFS) after treo- sulfan-based conditioning was evident. Furthermore, the treosulfan-based conditioning regimen in that study re- sulted in a lower TRM of 14% with an acceptable graft failure rate of 5%. Only the 11 patients receiving HCT from HLA partially matched family donors showed a poor outcome with a survival of only 9% compared with 73%
for matched related donor (MRD) and 63% for matched unrelated donor (MUD). The recent HLH-2004 study re- ported the results of HCT for children with HLH [48]. Of the total of 187 patients with HLH in that study, 134 had verified FHL and 99 received a conditioning regimen with busulfan-based, 39 with fludarabine-based and 20 with treosulfan-based. Donors were MRD in 44 patients, MUD in 60, haploidentical family donor (HFD) in 11, mis- matched unrelated donor (MMUD) in 6, and umbilical cord blood (UCB) in 53. The five-year OS was 66% but was 71% for verified FHL and 52% for nonverified HLH.
The EFS was 80% for treosulfan-based conditioning but this was not statistically different from the other con- ditioning regimens (59% for the busulfan-based and 50%
for the fludarabine-based regimens). The EFS for the pa- tients who received HCT from a HFD was 82% but this was not significantly from the cases with other types of donor (54% for MRD, 62% for MUD and 58% for UCB).
HCT is a curative treatment for patients with re- fractory and relapsed as well as familial HLH. While a human leukocyte antigen (HLA)-identical sibling donor is the preferred choice for HCT for HLH, a matched un- related volunteer donor is a realistic option for successful HCT. Unfortunately, significant proportion of patients with HLH requiring HCT cannot find an HLA-matched
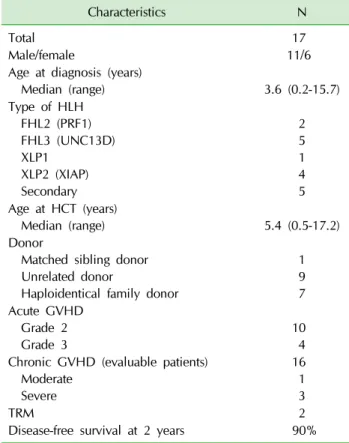
Table 3. Patient and transplant characteristics
Characteristics N
Total 17
Male/female 11/6
Age at diagnosis (years)
Median (range) 3.6 (0.2-15.7)
Type of HLH FHL2 (PRF1) FHL3 (UNC13D) XLP1
XLP2 (XIAP) Secondary
2 5 1 4 5 Age at HCT (years)
Median (range) 5.4 (0.5-17.2)
Donor
Matched sibling donor Unrelated donor
Haploidentical family donor
1 9 7 Acute GVHD
Grade 2
Grade 3 10
4 Chronic GVHD (evaluable patients)
Moderate Severe
16 1 3
TRM 2
Disease-free survival at 2 years 90%
related or unrelated donor. Alternatively, allogeneic HCT from a HFD could be a donor source for virtually all pa- tients who require HCT. Although the early experiences with HCT from a HFD were disappointing, recent ad- vances in T cell regulation strategies, including the ex vivo depletion of T cells and posttransplant cyclo- phosphamide (PTCY), have significantly improved the outcomes of haploidentical HCT [49-52]. The recent emerging evidence for haploidentical HSCT has provided additional therapeutic option to consider in pediatric patients with malignant and non-malignant disease cur- able with HSCT but who have not a suitable related or unrelated donor [53-56]. Medina-Valencia et al. reported a successful haploidentical HCT with PTCY in 5 pediatric patients with primary HLH [57]. Recently, an ex vivo T cell-depleted haploidentical HCT after treosulfan-based conditioning has been reported in 12 patients with fami- lial HLH [58]. The conditioning in those cases consisted of treosulfan, fludarabine, thiotepa and anti-thymocyte globulin. Four patients died, mostly due to infections, leading to a disease-free survival rate of 58.3%.
Experiences with Treosulfan-Based Conditioning for HLH at Asan Medical
Center Children’s Hospital (AMCCH), Unpublished Data
Between January 2016 and November 2020, 17 chil- dren and adolescents with HLH received HCT after treo- sulfan-based conditioning regimen at our hospital (AMCCH).
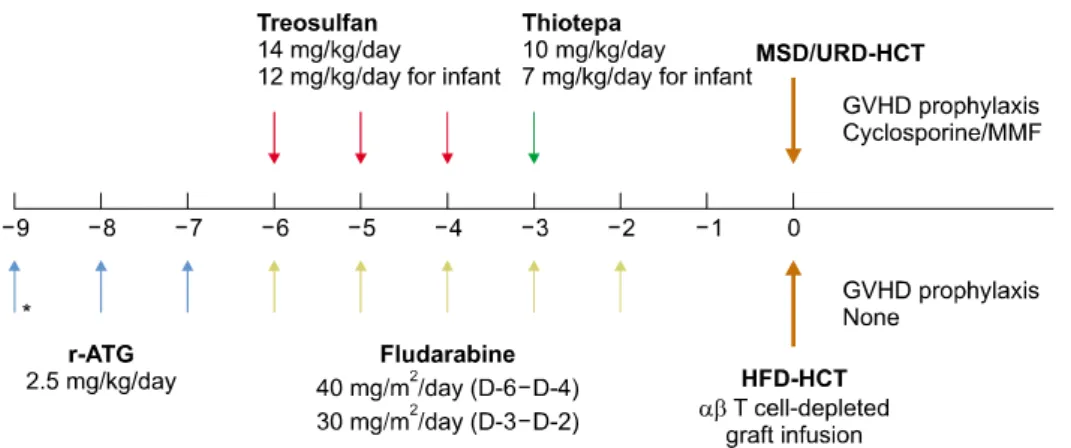
Patient and transplant characteristics are summarized in Table 3. Seven patients had familial HLH (2 PRF1 and 5 UNC13D), 5 had other genetic defects (4 XIAP and 1 XLP1) and 5 had secondary HLH with relapsed or re- fractory disease. Two patients with XIAP received HCT for intractable bowel disease prior to the appearance of HLH. The donors were a matched sibling donor (MSD) in 1 patient, an unrelated donor (URD) in 9 and a hap- loidentical family donor (HFD) in 7. The conditioning regimen consisted of treosulfan at a total dose of 42 mg/kg, except for 2 infants who received 36 mg/kg, flu- darabine at a total dose of 150-180 mg/m2, thiotepa at
a total dose of 7-10 mg/kg, and rabbit ATG (r-ATG, thy- moglobulin) at a total dose of 5-7.5 mg/kg, respectively (Fig. 2). In the patients receiving HCT from a MSD or URD, cyclosporine and mycophenolate mofetil (MMF) were administered for GVHD prophylaxis. For the hap- loidentical HCT, an ex vivo αβ T cell-depleted graft was used with no pharmacological GVHD prophylaxis. All 17 patients achieved sustained engraftment at a median of 10 days for neutrophils and 16 days for platelets, respectively. No patients experienced late graft failure.
Regimen-related toxicities were mild to moderate, and only one patient developed VOD which was successfully treated with defibrotide. Ten patients (59%) developed a grade 2 acute GVHD involving the skin only which was successfully treated with addition of systemic steroid.
Acute grade 3 GVHD occurred in 4 patients (24%) who were fully resolved by treatment with systemic steroid, infliximab and/or other immunosuppressants. No pa- tients developed acute grade 4 GVHD. Of the 16 evalu- able patients, 1 patient developed moderate chronic
Fig. 2. HCT for pediatric patients with HLH. *Additional dose of r-ATG is given for MSD- and URD- HCT. Two doses of r-ATG is ad- ministered in HFD-HCT. MSD, matched sibling donor; URD, un- related donor; HFD, haploidenti- cal family donor; MMF, mycopheno- late mofetil.
GVHD, and 3 patients developed severe chronic GVHD.
Among our 17 patients, 15 survived with complete do- nor chimerism and 2 patients died. One patient, who re- ceived HCT from a haploidentical mother, achieved neu- trophil engraftment at D+10, but the patient’s chimerism showed 83% from donor origin with declining neutrophil counts to 480 μL/mL. DLIs at a dose of 2.5×104 T cells/kg were given at D+47 and D+55 and full donor chimerism was achieved at D+61. The patient developed grade 3 acute GVHD involving the skin and gut after DLI, which was resolved by treatment with systemic steroids, tacroli- mus and infliximab. However, this patient died of pneu- monia at 24 months after HCT from HFD. The other de- ceased patient, who received URD-HCT, achieved neu- trophil engraftment at D+10 with full donor chimerism.
Severe chronic GVHD developed in the patient however at D+130 posttransplant for which systemic steroids, my- cophenolate mofetil and imatinib were administered.
While receiving this multiple immunosuppressants, the patient died of asphyxia at 35 months after HCT.
At a median follow-up of 25 months, the probabilities of overall survivals (OS) and disease-free survival (DFS) were same at 90±9.5% at 2 years. The 2-year probability of moderate-to-severe chronic GVHD-free and disease- free survival was 74±11.3%.
Our findings thus indicated that HCT using treosulfan/
fludarabine/thiotepa/r-ATG shows a favorable outcome with excellent donor chimerism in pediatric patients with HLH. Furthermore, haploidentical HCT from an αβ T cell-depleted graft could be a possible alternative in
patients who lack a matched related or unrelated donor.
The high incidence of acute GVHD evident in our study requires further modification of the conditioning regi- men or GVHD prophylaxis. Further studies are still need- ed to clarify and verify the safety and efficacy of treo- sulfan-based conditioning in pediatric patients with HLH.
Conclusion
Treosulfan-based conditioning improves the outcomes of HLH and is a well-established RTC regimen in children and adolescents with this disease as well as other non- malignant diseases. Notably however, further improve- ments to the conditioning regimen will be necessary to achieve full donor chimerism with an acceptable low rate of GVHD. For this purpose, a combination of other agents, personalized dosing of treosulfan based on phar- macokinetic data, and modifications to the GVHD pro- phylaxis including serotherapy could be considered. In addition, further study is warranted to evaluate the long-term toxicities and complications related to this RTC regimen.
Acknowledgments
This work was supported by the Research Program funded by the Korea Disease Control and Prevention Agency (2019-ER6903-02).
Conflict of Interest Statement
The authors have no conflict of interest to declare.
References
1. Janka GE. Familial hemophagocytic lymphohistiocytosis. Eur J Pediatr 1983;140:221-30.
2. Filipovich AH, Chandrakasan S. Pathogenesis of hemophago- cytic lymphohistiocytosis. Hematol Oncol Clin North Am 2015;29:895-902.
3. Risma KA, Marsh RA. Hemophagocytic lymphohistiocytosis:
clinical presentations and diagnosis. J Allergy Clin Immunol Pract 2019;7:824-32.
4. Zhang L, Zhou J, Sokol L. Hereditary and acquired hemopha- gocytic lymphohistiocytosis. Cancer Control 2014;21:301-12.
5. Scott Baker K, Jordan MB. Hematopoietic cell transplantation and novel therapies in hemophagocytic lymphohistiocytosis.
In: Abla O, Janka G, editors. Histiocytic disorders. New York:
Springer International Publishing, 2018;265-74.
6. Griffin G, Shenoi S, Hughes GC. Hemophagocytic lymphohis- tiocytosis: an update on pathogenesis, diagnosis, and therapy.
Best Pract Res Clin Rheumatol 2020;34:101515.
7. Degar B. Familial hemophagocytic lymphohistiocytosis. Hematol Oncol Clin North Am 2015;29:903-13.
8. Henter JI, Samuelsson-Horne A, Aricò M, et al. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immuno- chemotherapy and bone marrow transplantation. Blood 2002;
100:2367-73.
9. Trottestam H, Horne A, Arico M, et al. Chemoimmunotherapy for hemophagocytic lymphohistiocytosis: long-term results of the HLH-94 treatment protocol. Blood 2011;118:4577-84.
10. Durken M, Horstmann M, Bieling P, et al. Improved outcome in haemophagocytic lymphohistiocytosis after bone marrow transplantation from related and unrelated donors: a sin- gle-centre experience of 12 patients. Br J Haematol 1999;
106:1052-8.
11. Ehl S, Astigarraga I, von Bahr Greenwood T, et al. Recom- mendations for the use of etoposide-based therapy and bone marrow transplantation for the treatment of HLH: consensus statements by the HLH steering Committee of the Histiocyte Society. J Allergy Clin Immunol Pract 2018;6:1508-17.
12. Horne A, Janka G, Maarten Egeler R, et al. Haematopoietic stem cell transplantation in haemophagocytic lymphohistio- cytosis. Br J Haematol 2005;129:622-30.
13. Marsh RA, Vaughn G, Kim MO, et al. Reduced-intensity con- ditioning significantly improves survival of patients with he- mophagocytic lymphohistiocytosis undergoing allogeneic hem- atopoietic cell transplantation. Blood 2010;116:5824-31.
14. Marsh RA, Jordan MB, Filipovich AH. Reduced-intensity con- ditioning haematopoietic cell transplantation for haemopha- gocytic lymphohistiocytosis: an important step forward. Br J Haematol 2011;154:556-63.
15. Danylesko I, Shimoni A, Nagler A. Treosulfan-based condition- ing before hematopoietic SCT: more than a BU look-alike.
Bone Marrow Transplant 2012;47:5-14.
16. ten Brink MH, Zwaveling J, Swen JJ, Bredius RG, Lankester AC, Guchelaar HJ. Personalized busulfan and treosulfan con- ditioning for pediatric stem cell transplantation: the role of pharmacogenetics and pharmacokinetics. Drug Discov Today 2014;19:1572-86.
17. Seger RA, Gungor T, Belohradsky BH, et al. Treatment of ch- ronic granulomatous disease with myeloablative conditioning and an unmodified hemopoietic allograft: a survey of the European experience, 1985-2000. Blood 2002;100:4344-50.
18. Bernaudin F, Socie G, Kuentz M, et al. Long-term results of related myeloablative stem-cell transplantation to cure sickle cell disease. Blood 2007;110:2749-56.
19. Ghavamzadeh A, Iravani M, Ashouri A, et al. Peripheral blood versus bone marrow as a source of hematopoietic stem cells for allogeneic transplantation in children with class I and II beta thalassemia major. Biol Blood Marrow Transplant 2008;
14:301-8.
20. Baker KS, Filipovich AH, Gross TG, et al. Unrelated donor hematopoietic cell transplantation for hemophagocytic lym- phohistiocytosis. Bone Marrow Transplant 2008;42:175-80.
21. Cesaro S, Locatelli F, Lanino E, et al. Hematopoietic stem cell transplantation for hemophagocytic lymphohistiocytosis: a retrospective analysis of data from the Italian Association of Pediatric Hematology Oncology (AIEOP). Haematologica 2008;
93:1694-701.
22. Ohga S, Kudo K, Ishii E, et al. Hematopoietic stem cell trans- plantation for familial hemophagocytic lymphohistiocytosis and Epstein-Barr virus-associated hemophagocytic lympho- histiocytosis in Japan. Pediatr Blood Cancer 2010;54:299-306.
23. Yoon HS, Im HJ, Moon HN, et al. The outcome of hema- topoietic stem cell transplantation in Korean children with hemophagocytic lymphohistiocytosis. Pediatr Transplant 2010;
14:735-40.
24. Cooper N, Rao K, Gilmour K, et al. Stem cell transplantation with reduced-intensity conditioning for hemophagocytic lym- phohistiocytosis. Blood 2006;107:1233-6.
25. Allen CE, Marsh R, Dawson P, et al. Reduced-intensity con- ditioning for hematopoietic cell transplant for HLH and pri- mary immune deficiencies. Blood 2018;132:1438-51.
26. Cooper N, Rao K, Goulden N, Webb D, Amrolia P, Veys P.
The use of reduced-intensity stem cell transplantation in haemophagocytic lymphohistiocytosis and Langerhans cell histiocytosis. Bone Marrow Transplant 2008;42 Suppl 2:S47- 50.
27. Marsh RA, Kim MO, Liu C, et al. An intermediate alemtuzu- mab schedule reduces the incidence of mixed chimerism fol- lowing reduced-intensity conditioning hematopoietic cell transplantation for hemophagocytic lymphohistiocytosis. Biol Blood Marrow Transplant 2013;19:1625-31.
28. Gropp M, Meier W, Hepp H. Treosulfan as an effective sec- ond-line therapy in ovarian cancer. Gynecol Oncol 1998;71:
94-8.
29. Schmidmaier R, Oellerich M, Baumgart J, Emmerich B, Meinhardt G. Treosulfan-induced apoptosis in acute myeloid
leukemia cells is accompanied by translocation of protein kinase C delta and enhanced by bryostatin-1. Exp Hematol 2004;32:76-86.
30. Munkelt D, Koehl U, Kloess S, et al. Cytotoxic effects of treo- sulfan and busulfan against leukemic cells of pediatric pa- tients. Cancer Chemother Pharmacol 2008;62:821-30.
31. Westerhof GR, Ploemacher RE, Boudewijn A, et al. Comparison of different busulfan analogues for depletion of hemato- poietic stem cells and promotion of donor-type chimerism in murine bone marrow transplant recipients. Cancer Res 2000;
60:5470-8.
32. Ploemacher RE, Johnson KW, Rombouts EJ, et al. Addition of treosulfan to a nonmyeloablative conditioning regimen re- sults in enhanced chimerism and immunologic tolerance in an experimental allogeneic bone marrow transplant model.
Biol Blood Marrow Transplant 2004;10:236-45.
33. Sjoo F, Hassan Z, Abedi-Valugerdi M, et al. Myeloablative and immunosuppressive properties of treosulfan in mice. Exp Hematol 2006;34:115-21.
34. Greystoke B, Bonanomi S, Carr TF, et al. Treosulfan-contain- ing regimens achieve high rates of engraftment associated with low transplant morbidity and mortality in children with non-malignant disease and significant co-morbidities. Br J Haematol 2008;142:257-62.
35. Slatter MA, Rao K, Amrolia P, et al. Treosulfan-based con- ditioning regimens for hematopoietic stem cell transplan- tation in children with primary immunodeficiency: United Kingdom experience. Blood 2011;117:4367-75.
36. Burroughs LM, Nemecek ER, Torgerson TR, et al. Treosulfan- based conditioning and hematopoietic cell transplantation for nonmalignant diseases: a prospective multicenter trial.
Biol Blood Marrow Transplant 2014;20:1996-2003.
37. Dinur-Schejter Y, Krauss AC, Erlich O, et al. Bone marrow transplantation for non-malignant diseases using treosulfan- based conditioning. Pediatr Blood Cancer 2015;62:299-304.
38. Slatter MA, Boztug H, Potschger U, et al. Treosulfan-based conditioning regimens for allogeneic haematopoietic stem cell transplantation in children with non-malignant diseases.
Bone Marrow Transplant 2015;50:1536-41.
39. Morillo-Gutierrez B, Beier R, Rao K, et al. Treosulfan-based conditioning for allogeneic HSCT in children with chronic granulomatous disease: a multicenter experience. Blood 2016;
128:440-8.
40. Burroughs LM, Shimamura A, Talano JA, et al. Allogeneic hematopoietic cell transplantation using treosulfan-based conditioning for treatment of marrow failure disorders. Biol Blood Marrow Transplant 2017;23:1669-77.
41. Slatter MA, Rao K, Abd Hamid IJ, et al. Treosulfan and fludar- abine conditioning for hematopoietic stem cell transplan- tation in children with primary immunodeficiency: UK ex- perience. Biol Blood Marrow Transplant 2018;24:529-36.
42. Naik S, Eckstein O, Sasa G, et al. Incorporation of thiotepa in a reduced intensity conditioning regimen may improve en- graftment after transplant for HLH. Br J Haematol 2020;188:
e84-7.
43. Glowka FK, Karazniewicz-Lada M, Grund G, Wrobel T, Wachowiak J. Pharmacokinetics of high-dose i.v. treosulfan
in children undergoing treosulfan-based preparative regimen for allogeneic haematopoietic SCT. Bone Marrow Transplant 2008;42 Suppl 2:S67-70.
44. Romanski M, Wachowiak J, Glowka FK. Treosulfan pharma- cokinetics and its variability in pediatric and adult patients undergoing conditioning prior to hematopoietic stem cell transplantation: current state of the art, in-depth analysis, and Perspectives. Clin Pharmacokinet 2018;57:1255-65.
45. Lehmberg K, Albert MH, Beier R, et al. Treosulfan-based conditioning regimen for children and adolescents with he- mophagocytic lymphohistiocytosis. Haematologica 2014;99:
180-4.
46. Wustrau K, Greil J, Sykora KW, et al. Risk factors for mixed chimerism in children with hemophagocytic lymphohistiocy- tosis after reduced toxicity conditioning. Pediatr Blood Can- cer 2020;67:e28523.
47. Messina C, Zecca M, Fagioli F, et al. Outcomes of children with hemophagocytic lymphohistiocytosis given allogeneic hematopoietic stem cell transplantation in Italy. Biol Blood Marrow Transplant 2018;24:1223-31.
48. Bergsten E, Horne A, Hed Myrberg I, et al. Stem cell trans- plantation for children with hemophagocytic lymphohistio- cytosis: results from the HLH-2004 study. Blood Adv 2020;
4:3754-66.
49. Lang P, Feuchtinger T, Teltschik HM, et al. Improved immune recovery after transplantation of TCRalphabeta/CD19-de- pleted allografts from haploidentical donors in pediatric patients. Bone Marrow Transplant 2015;50 Suppl 2:S6-10.
50. Im HJ, Koh KN, Suh JK, et al. Haploidentical HCT using an alphabeta T-cell-depleted graft with targeted alphabeta(+) cells by add-back after a reduced intensity preparative regi- men containing low-dose TBI. Bone Marrow Transplant 2016;
51:1217-22.
51. Locatelli F, Merli P, Pagliara D, et al. Outcome of children with acute leukemia given HLA-haploidentical HSCT after al- phabeta T-cell and B-cell depletion. Blood 2017;130:677-85.
52. Symons HJ, Zahurak M, Cao Y, et al. Myeloablative haplo- identical BMT with posttransplant cyclophosphamide for hematologic malignancies in children and adults. Blood Adv 2020;4:3913-25.
53. Bertaina A, Merli P, Rutella S, et al. HLA-haploidentical stem cell transplantation after removal of alphabeta+ T and B cells in children with nonmalignant disorders. Blood 2014;124:
822-6.
54. Uppuluri R, Sivasankaran M, Patel S, et al. Haploidentical stem cell transplantation with post-transplant cyclopho- sphamide for primary immune deficiency disorders in chil- dren: challenges and outcome from a tertiary care center in South India. J Clin Immunol 2019;39:182-7.
55. Fernandes JF, Nichele S, Arcuri LJ, et al. Outcomes after hap- loidentical stem cell transplantation with post-transplan- tation cyclophosphamide in patients with primary immuno- deficiency diseases. Biol Blood Marrow Transplant 2020;26:
1923-9.
56. Torres Canizales J, Ferreras C, Pascual A, et al. Haploidentical transplantation in pediatric non-malignant diseases: a retro- spective analysis on behalf of the Spanish Group for Hemato-
poietic Transplantation (GETH). Eur J Haematol 2021;106:
196-204.
57. Medina-Valencia D, Cleves D, Beltran E, et al. Haploidentical stem cell transplant with post-transplant cyclophosphamide in pediatric hemophagocytic lymphohistiocytosis. J Clin Immunol 2021.
58. Nazir HF, Ba Alawi FS, Al Hosni S, Al Rawas A, Dennison D.
T cell depleted haploidentical hematopoietic stem cell trans- plantation for patients with familial hemophagocytic lym- phohistiocytosis who do not have matched family donors:
experience in Oman. Biol Blood Marrow Transplant 2020;26:
1119-23.