Application of Polyaniline to an Enzyme-Amplified Electrochemical Immunosensor
as an Electroactive Report Molecule
††Seong Jung Kwon, Myungeun Seo, Haesik Yang,‡ Sang Youl Kim, and Juhyoun Kwak*
Department of Chemistry, Korea Advanced Institute of Science and Technology (KAIST), Daejeon 305-701, Korea *E-mail: [email protected]
‡Department of Chemistry and Chemistry Institute for Functional Materials, Pusan National University, Busan 609-735, Korea
Received July 6, 2010, Accepted July 16, 2010
Conducting polymers (CPs) are widely used as matrixes for the entrapment of enzymes in analytical chemistry and biosensing devices. However, enzyme-catalyzed polymerization of CPs is rarely used for immunosensing due to the difficulties involved in the quantitative analysis of colloidal CPs in solution phase. In this study, an enzyme-amplified electrocatalytic immunosensor employing a CP as a redox marker has been developed. A polyanionic polymer matrix, α-amino-ω-thiol terminated poly(acrylic acid), was employed for precipitation of CP. The acrylic acid group acts as a polyanionic template. The thiol terminus of the polymer was used to produce self-assembled monolayers (SAMs) on Au electrodes and the amine terminus was employed for immobilization of biomolecules. In an enzyme- amplified sandwich type immunosensor, the polyaniline (PANI) produced enzymatically is attracted by the electro-static force of the matrix polymer. The precipitated PANI was characterized by electrochemical methods. Key Words: Polyaniline, Conducting polymer, Enzymatic synthesis, Electrochemical detection, Immunosensor
Introduction
An immunosensor is a device having an antibody-antigen sensing element and a transducer.1 The detection systems for immunosensors are mainly based on optical, electrochemical, or gravimetric methods.2,3 In the case of electrochemical immuno-sensors, the antibody-antigen interaction is transformed into an electrical signal.4,5 An electroactive molecule that is used as a label is referred to as a redox marker. The signal can be ampli-fied by an enzyme that continuously generates redox markers.6 Because of their simple fabrication process and good sensi-tivity, enzyme-amplified electrochemical immunosensors have been used widely.7-10 Redox markers produced by an enzyme are usually small organic or inorganic molecules. However, conducting polymers (CPs) are a good substitute for the redox marker in biosensors because of their stable redox reaction and good biocompatibility. In addition to application in electro-chemical detection, they also can be used for optical detection due to their optical properties.
CPs have π-electron backbones, which are responsible for their unusual electronic properties including electrical conduc-tivity, low energy optical transitions, high electron affinity, and low ionization potential. Providing the possibility of surface modification of conventional electrodes, CPs have been widely used in analytical chemistry and biosensing devices.11-13 They have also attracted interest as a suitable matrix for the entrap-ment of enzymes.14,15 Notably, CPs can act as a redox marker in an enzyme amplified electrochemical biosensor because they are electroactive enzyme products. In addition to electrochemi-cal and chemielectrochemi-cal methods, the synthesis of polymers through enzymatic polymerization has been extensively developed.16,17 †This paper is dedicated to Professor Hasuck Kim for his outstanding contribution to electrochemistry and analytical chemistry.
This method is recognized as a new area of precision polymer synthesis. Horseradish peroxidase (HRP) is capable of catalyz-ing the polymerization of anilines and phenols in the presence of hydrogen peroxide to generate their respective free radicals. These free radicals undergo coupling to produce dimers. Su-ccessive oxidation and coupling reactions result in polymer formation.18-21
The use of a CP as a redox marker requires specific condi-tions such as a polyanionic template for growing the polymer and an acidic pH condition for maintaining conductivity. A quantitative analysis of colloidal CP in a solution phase is difficult, and thus precipitation on the electrode surface is also necessary. Therefore, the applications of CP as a redox marker have been reported only in DNA sensor to date.22,23 In these studies, the negatively charged DNA backbone itself played the role of a polyanionic template to precipitate CP. However, the precipitation of CP is difficult to realize in an immuno-sensor in the absence of a polyanionic template. In this paper, HRP-catalyzed polymerization was used as an enzyme reaction for immunosensing. We employed a polyanionic polymer mat-rix, α-amino-ω-thiol terminated poly(acrylic acid), which has multi-functional groups in its chains. The thiol termini were used for self-assembled monolayers on an Au electrode. The amine termini were used for immobilization of a biomolecule and the acrylic acid group acts as a polyanionic template. In a sandwich type immunosensor, the labeled HRP enzyme produces poly-aniline (PANI) and it is attracted by the electrostatic force of the matrix polymer and thereupon precipitated. The produced PANI layer can be characterized by electrochemical methods.
Experimental
HRP-conjugated goat antimouse IgG, mouse antibiotin IgG, streptavidin, and soybean peroxidase (SBP) were purchased from Sigma. Aniline and p-toluene sulfonic acid (p-TSA) (so-dium salt) were purchased from Aldrich. 1-ethyl-3-(3-dimethyl-aminopropyl)carbodiimide hydrochloride (EDC), 3,3'-Dithiobis (sulfosuccinimidylpropionate) (DTSSP), sulfo-N-hydroxy-succinimide (NHS)-biotin, amine-poly ethylene oxide (PEO)- biotin, and biotinylated goat antimouse IgG were obtained from Pierce (Rockford, IL). Hydrogen peroxide was purchased from Junsei. α-Amino-ω-thiol terminated poly(t-butyl acrylate) (Mn = 8.8 × 103, PDI = 1.09) was purchased from Polymer Source, Inc. (Montreal, Canada) and used without further purification. Trifluoroacetic acid was purchased from TCI. All buffer salts and other inorganic chemicals were obtained from Sigma or Aldrich, unless otherwise stated. All chemicals were used as received. Ultra-pure water (> 18 MΩ, Millipore) was used in all experiments. The incubating buffer (IB) consisted of 50 mM Tris, 150 mM NaCl, and 1% bovine serum albumin (BSA) (pH 7.2). The rinsing buffer (RB) was comprised of 50 mM Tris, 0.5 M NaCl, 0.05% Tween 20, and 0.05% BSA (pH 7.5).
Hydrolysis of α-amino-ω-thiol terminated poly(t-butyl acryl-ate). α-Amino-ω-thiol terminated poly(acrylic acid) was syn-thesized by hydrolysis of the t-butyl group of α-amino-ω-thiol terminated poly(t-butyl acrylate). To a solution of α-amino-ω- thiol terminated poly(t-butyl acrylate) (0.4 g) in dichloromethane (20 mL), trifluoroacetic acid (2.5 mL) was added and stirred for 36 h at room temperature. The reaction mixture was poured into an excess of n-hexane and the crude product was obtained by filtration. The crude product was dissolved in DMSO and purified by dialysis in a 1 wt % triethylamine aqueous solution for 48 h using a Spectra/Por 3 dialysis membrane (molecular weight cutoff ~3500). Evaporation of water yielded the pro-duct (0.21 g), which was then dissolved in 10 mL of water to make a stock solution.
Preparation of an immunosensing layer.
Preparation of enzyme immobilized electrode: Gold elect-rodes were prepared by electron-beam evaporation of 40 nm of Ti followed by 150 nm of Au onto Si (100) wafers. The elect-rode was cleaned in a piranha solution [H2O2 : H2SO4 (v/v) = 1 : 3], rinsed with water, and dried under N2 gas. The cleaned Au electrode was immersed in a solution of 5 mM DTSSP for 12 h, and then rinsed with water. The activated succinimide groups were reacted with the primary amine of SBP by immer-sion in IB containing 1 mg/mL SBP for 1 h and then washed with RB.
Preparation of an enzyme-linked immunosorbent assay (ELISA) type immunosensor: The cleaned Au electrode was immersed in a solution of 5 mM DTSSP for 12 h, and then washed with water. The activated succinimide groups were reacted with the primary amine by immersion of the electrode into a solution of 25 mM amine-PEO-biotin for 3 h, followed by washing with water. The biotmodified electrode was in-cubated in IB for 30 min to prevent non-specific adsorption of proteins, and then washed with RB. The biotinylated electrode was immersed in IB containing 100 µg/mL of mouse antibiotin IgG for 30 min. After washing with RB, the electrode was finally incubated with 100 µg/mL of HRP-conjugated goat antimouse IgG for 40 min and then washed with RB.
Preparation of sandwich type immunosensor: The cleaned Au electrode was immersed in a stock solution of α-amino-ω- thiol terminated poly(acrylic acid) for 12 h, and then washed with water. The amine groups were reacted with NHS by immer-sion of the electrode into a solution of 10 mM sulfo-NHS-biotin and 50 mM EDC for 3 h, followed by washing with water. The biotin-modified electrode was incubated in IB for 30 min to prevent non-specific adsorption of proteins, and then washed with RB. The electrode was immersed in IB containing 100 µg/ mL of streptavidin for 1 h. After rinsing with RB, the resultant assembly was immersed in IB containing 100 µg/mL of bio-tinylated goat antimouse IgG for 30 min. After washing with RB, various concentrations of target mouse IgG in IB were captured by the immunosensor for 30 min, followed by washing with RB. The immunosensor was finally incubated with 100 µg/mL of HRP-conjugated goat antimouse IgG for 40 min and then washed with RB.
Enzymatic polymerization. The biosensing layer with HRP modified on the surface was incubated for 3 h in a 2-(N-mor-pholino)ethanesulfonic acid (MES) (pH 5.5) buffer containing 10 mM aniline monomer, 10 mM hydrogen peroxide, and 10 mM p-TSA. The 10 mM aliquot of p-TSA was absent in the sandwich type immunosensor. According to enzymatic poly-merization, a dark brown product, PANI, was precipitated. This was rinsed twice with RB. The electrode was then moved into a solution containing 0.1 M H2SO4 for electrochemistry.
Electrochemical experiment and instruments. The chemical experiment was performed using a BAS 100B electro-chemical analyzer (Bioanalytical Systems, Inc.). The three-elect-rode electrochemical cell consisted of a modified gold working electrode (area: 0.283 cm2), a Pt wire counter electrode, and a silver/silver chloride reference electrode (Ag/AgCl electrode containing saturated 3 M NaCl). 1H NMR spectra of the com-pounds were obtained using a Bruker 300 MHz spectrometer with the residual solvent signal as an internal reference.
Results and Discussion
Polyaniline (PANI) is one of the most stable conducting polymers and also one of the easiest to synthesize. It displays good conductivity in combination with high stability in its oxi-dized form. The redox properties of PANI are affected by the solvent or the cation of the electrolyte. Their conductivity is changed according to oxidation states or doping level.24,25 To identify the redox potential of PANI, it was prepared by electro-polymerization. Generally, electropolymerization of CP is per-formed in a strong acid solution. However, a pH 5.5 buffer was used here for comparison with the enzyme-synthesized product. The enzyme is easily denatured according to temperature or pH change. For HRP in particular, the optimal pH is around pH 6.5.26 Therefore, the enzymatic polymerization and electro-chemical characterization steps should be separated for effec-tive production and doping. As shown in the inset of Figure 1a, a redox peak at 0.50 V (vs. Ag/AgCl) appeared and was in-creased by cycling. PANI is responsible for this peak and the small incremental change indicates the electrode surface is blocked by less-conductive PANI, which is synthesized in a MES buffer (pH 5.5) containing 10 mM aniline monomer. PANI
0 400 800 E (mV) vs. Ag/AgCl ‒100 ‒80 ‒60 ‒40 ‒20 0 20 C u rre n t ( A ) μ A (a) 3rd scan 2nd scan 1st scan ‒200 0 200 400 600 800 E (mV) vs. Ag/AgCl ‒10 0 10 20 C u rre n t ( A ) μ A (b)
Figure 1. (a) Cyclic voltammograms for electropolymerization of poly-aniline in a MES buffer (pH 5.5) containing 10 mM poly-aniline monomer. (b) Cyclic voltammograms for redox reaction of electropolymerized polyaniline layer in a 0.1 M H2SO4 solution. Both scan rates are 20 mV/s. ‒200 0 200 400 600 800 E (mV) vs. Ag/AgCl (a) ‒4 ‒2 0 2 4 C u rre n t ( A ) μ A 0 100 200 300 400 500 600 Scan rate (mV/s) (b) ‒30 ‒20 ‒10 0 Ip ( A ) μ A
Figure 2. (a) Cyclic voltammograms for redox reaction of enzymati-cally synthesized polyaniline layer. Electrolyte is 0.1 M H2SO4 solu-tion. Scan rate is 20 mV/s. (b) Plot of anodic peak currents to potential sweep rates.
has good conductivity in acidic pHs, which is related with the oxidation state and doping level. As shown in Figure 1b, the current of the redox peak was increased in an electrolyte solu-tion containing 0.1 M H2SO4 due to the higher doping level in acid solution. Also, two more peaks, at 0.20 V and 0.75 V (vs. Ag/AgCl), which represent different oxidation states of surface- bound PANI, appeared after doping. Three idealized oxidation states of PANI, leucoemeraldin, emeraldine, and pernigraniline, were previously reported.27 The three peaks appearing in the acidic condition may represent these states.
To identify the redox potential of enzymatically polymerized PANI and to compare it with that of the electropolymerized product, an enzyme-attached electrode was prepared. SBPs were immobilized on a DTSSP SAM-modified electrode. SBP has better stability and activity than HRP in acidic pHs, and therefore was used here for higher yield.28-30 The dithiol bond in DTSSP was used to form SAMs on the Au electrode. The succinimide group reacts with the primary amine of SBP to form covalent bonds. The electrode was then investigated in terms of the enzymatic polymerization. The SBP-modified surface was incubated for 3 h in a MES buffer (pH 5.5) containing 10 mM aniline monomer, 10 mM hydrogen peroxide, and 10 mM
p-TSA. The deposited PANI on the electrode was electrochemi-cally characterized after being moved to a 0.1 M H2SO4 solu-tion. As shown in Figure 2a, two peaks appeared, at ca. 0.25 V and 0.55 V (vs. Ag/AgCl). They were assigned as ca. 0.05 V positive shifts of the peaks at 0.20 V and 0.50 V for the electro-polymerized PANI. It appears that the 0.05 V potential shift can be attributed to the additional layer of DTSSP SAMs and enzymes. It is determined that these two peak are from the de-posited PANI on the electrode surface, because the peak current was proportional to the scan rate, as shown in Figure 2b. The redox potential of enzymatically produced PANI on the elect-rode surface agrees closely with that of the electropolymerized PANI.
To test the possibility of utilizing CP as a redox marker in an immunosensor, we prepared an ELISA-type immunosensing layer. After making DTSSP SAMs on a gold electrode, biotin- NHS, antibiotin IgG, and HRP conjugated antimouse IgG were immobilized sequentially. In the presence of a high concent-ration of antibiotin IgG, we could see a distinguishable signal, as shown in Figure 3. HRP is an effective catalyst for oxidative polymerization of aniline in the presence of hydrogen peroxide at room temperature. Peroxidase-catalyzed polymerization
pro-Scheme 1. Synthesis of α-amino-ω-thiol terminated poly(acrylic acid) by hydrolysis of t-butyl group of α-amino-ω-thiol terminated poly (t-butyl acrylate)
Aniline
PANI Enzyme labeled IgG
Target IgG Biotinylated probe IgG
Avidin
Polymer
Scheme 2. Schematic illustration of a sandwich-type electrochemical immunosensor employing PANI as an electroactive report molecule ‒200 0 200 400 600 800 E (mV) vs. Ag/AgCl ‒2 ‒1 0 1 2 C u rre n t ( A ) μ A
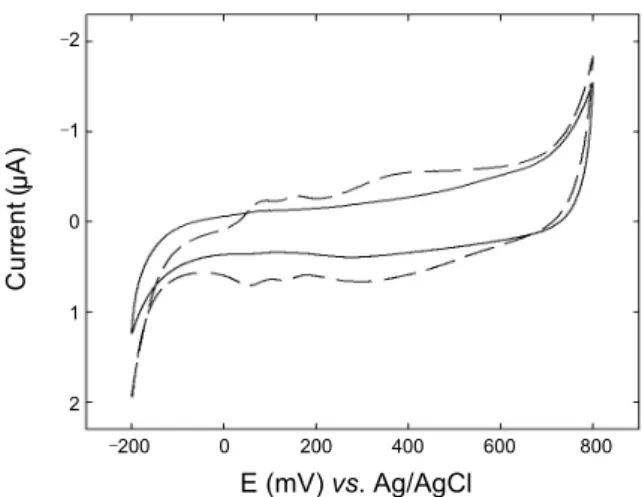
Figure 3. Cyclic voltammograms for redox reaction of enzymatically synthesized polyaniline precipitation in ELISA type immunosensor, with (dashed) and without (solid) an antibiotin IgG layer. Electrolyte is 0.1 M H2SO4 solution. Scan rate is 20 mV/s.
‒200 0 200 400 600 800 E (mV) vs. Ag/AgCl ‒8 ‒6 ‒4 ‒2 0 2 4 6 8 C u rre n t ( A ) μ A
Figure 4. Cyclic voltammograms for redox reaction of enzymatically synthesized polyaniline precipitation on the α-amino-ω-thiol termi-nated poly(acrylic acid) modified electrode before (solid) and after (dashed) being immersed for 30 min in a solution containing colloidal polyaniline. The colloidal polyaniline was enzymatically polymerized in a MES buffer (pH 5.5) containing 100 µg/mL of HRP, 10 mM aniline monomer, 10 mM H2O2, and 10 mM p-TSA for 10 min. Electrolyte is 0.1 M H2SO4 solution. Scan rate is 50 mV/s.
duces low-molecular-weight products. Therefore, the use of an anionic polyelectrolyte as a template for polymer growth is critical in polymerization. However, the synthesized PANI did not appear to precipitate spontaneously and most of it was washed out during the rinsing step, even though a polyanionic template was present in the solution. This indicates that the colloidal PANI in the solution phase is difficult to detect with electrochemical methods. Hence, precipitation and accumula-tion of synthesized PANI are necessary for more sensitive immunosensing.
To induce precipitation, we employed α-amino-ω-thiol ter-minated poly(acrylic acid) as a matrix for the immunosensor. The α-amino-ω-thiol terminated poly(acrylic acid) was prepared by hydrolysis of the t-butyl group of α-amino-ω-thiol terminated poly(t-butyl acrylate), as illustrated in Scheme 1. This provides
multiple functions. The thiol terminus is used for making SAMs on the gold electrode. The surface covered polymer matrix pre-vents denaturing of the attached enzymes on the electrode and helps enzymes retain their stability and activity. Also, the orga-nic polymer layer reduces the background current and non- specific binding. The primary amine terminus at the opposite side is used for immobilization of a biomolecule using EDC/ NHS coupling. The many acrylate groups in the polymer body provide negative charges to act as polyanionic templates. They attract the cation radicals of aniline or small fragments of PANI and induce precipitation. To test the interaction between PANI and the polyanionic polymer SAMs modified electrode, the latter was immersed in a solution containing colloidal PANI, which was enzymatically synthesized in a solution phase. As shown in Figure 4, the polyanionic polymer SAM-modified electrode shows good attraction with the colloidal state of PANI. The redox potential shift in an acidic solution also occurred in this case. This is likely due to the additional layer of the poly-mer-PANI complex.
Scheme 2 illustrates the sandwich type immunosensing sys-tem developed in the current study. For this syssys-tem, the α-amino- ω-thiol terminated poly(acrylic acid) SAM-modified electrode was incubated sequentially in a solution containing sulfo-NHS- biotin, streptavidin, biotinylated antimouse IgG, target mouse IgG, and HRP-conjugated antimouse IgG. To characterize the electrocatalytic performance of the biosensing layer, the im-munosensor was incubated for 3 h in a MES buffer (pH 5.5) con-taining 10 mM hydrogen peroxide and 10 mM aniline monomer.
0 200 400 600 800 1000 E (mV) vs. Ag/AgCl (a) ‒30 ‒20 ‒10 0 10 20 30 C u rre n t ( A ) μ A 0.1 1 10 100 Mouse IgG conc. (μg/mL)
(b) 25 20 15 10 5 0 Nor m al iz e d an od ic cha rg e
Figure 5. (a) Cyclic voltammograms for redox reaction of enzymatic synthesized polyaniline precipitation in a sandwich type sensor with 0 g/mL (solid), 100 ng/mL (dotted), 1 µg/mL (dashed), 10 µg/mL (dash dotted), and 100 µg/mL (long dashed) of target mouse IgG concent-ration. Electrolyte is 0.1 M H2SO4 solution. Scan rate is 400 mV/s (b) Normalized anodic charge of each peak in Figure 5a. The charge is calculated in a range from 0 V to 1.0 V. The curve of 0 g/mL of target mouse IgG is used as a baseline.
The enzymatically polymerized CP is attracted by the acrylate groups of the matrix polymer and preferentially aligns the mono-mers through electrostatic and hydrophobic interactions to pro-mote the desired head-to-tail coupling. The electrode was rinsed to remove non-precipitated colloidal CP in a solution phase and was moved to an electrolyte cell containing 0.1 M H2SO4 for electrochemistry. Cyclic voltammograms of immunosensing electrodes were obtained at various concentrations of mouse IgG, as shown in Figure 5a. According to the concentration of target mouse IgG, the current at 0.5 V (vs. Ag/AgCl) was in-creased. We obtained single broad peak rather than two or three peaks. Above mentioned, number of peaks and redox potential of PANI can be changed due to the synthetic methods, counter anion, and surface modification of electrode. Though an accu-rate estimation of potential shift is difficult because of a diffe-rent counter anion and surface state in immunosensor, the broad peak obtained in Figure 5a can be regarded as a PANI redox reaction according the trend shown in Figure 1b, 2a, 3 and 4. The anodic charge of each peak in the cyclic voltammograms
was plotted to determine the detection limit for mouse IgG (Figure 5b). The detection limit is 100 ng/mL of target mouse IgG and the detection range is from 100 ng/mL to 100 µg/mL. At higher concentrations, it appears that the enzyme may be buried in the PANI precipitation and lose its activity.
Conclusion
In this study, an enzyme-amplified electrocatalytic immuno-sensor employing PANI as a redox marker has been developed. Using a polyanionic polymer matrix, α-amino-ω-thiol termi-nated poly(acrylic acid), enzymatically produced PANI was precipitated on an electrode. In a sandwich type immunosensor, the amount of produced PANI was increased according to the target mouse IgG and characterized by electrochemical me-thods. The detection limit was 100 ng/mL for target mouse IgG. Acknowledgments. This work was supported by the Nano/ Bio Science & Technology Program (2010-0008213) of the Mi-nistry of Education, Science and Technology and also supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (2010-0001951). It was also partially supported by a Korea Research Foundation Grant funded by the Korean Government (KRF-2008-314-C00234 and F01-2008-000-10153-0).
References
1. Gerard, M.; Chauey, A.; Malhotra, B. D. Biosens. Bioelectron. 2002, 17, 345.
2. Ghindilis, L.; Atanasov, P.; Wilkins, M.; Wilkins, E. Biosens.
Bio-electron. 1998, 13, 113.
3. Van Emon, J. M.; Gerlach, C. L.; Bowman, K. J. Chromatogr. B 1998, 715, 211.
4. Limoges, B.; Saveant, J.; Yazidi, D. J. Am. Chem. Soc. 2003, 125, 9192.
5. Kim, H.; Zhang, Y.; Heller, A. Anal. Chem. 2004, 76, 2411. 6. Patolsky, F.; Lichtenstein, A.; Willner, I. Nat. Biotechnol. 2001,
19, 253.
7. Wilson, M. S. Anal. Chem. 2005, 77, 1496. 8. Ehrnstrom, R. Lab Chip 2002, 2, 26N.
9. Kwon, S. J.; Kim, E.; Yang, H.; Kwak, J. Analyst 2006, 131, 402. 10. Kwon, S. J.; Yang, H.; Jo, K.; Kwak, J. Analyst 2008, 133, 1599. 11. Situmorang, M.; Gooding, J. J.; Hibbert, D. B.; Barnett, D. Biosens.
Bioelectron. 1998, 13, 953.
12. Trojanowicz, M.; Vel Krawczyk, T. K. Mikrochim. Acta 1995,
121, 167.
13. Schuhmann, W. Mikrochim. Acta 1995, 121, 1. 14. Adeloju, S. B.; Wallace, G. G Analyst 1996, 121, 699. 15. Sung, W. J.; Bae, Y. H. Anal. Chem. 2000, 72, 2177.
16. Xu, P.; Singh, A.; Kaplan, D. L. Adv. Polym. Sci. 2006, 194, 69. 17. Nissum, M.; Schiodt, C. B.; Welinder, K. G. Biochim. Biophy. Acta
2001, 1545, 339.
18. Nabid, M. R.; Entezami, A. A. J. App. Poly. Sci. 2004, 94, 254. 19. Won, K.; Kim, Y. H.; An, E. S.; Lee, Y. S.; Song, B. K.
Biomacro-molecules 2004, 5, 1.
20. Malinauskas, A.; Malinauskiene, J.; Ramanavicius, A.
Nanotech-nology 2005, 16, R51.
21. Kobayashi, S.; Uyama, H.; Kimura, S. Chem. Rev. 2001, 101, 3793. 22. Gao, Z.; Rafea, S.; Lim, L. H. Adv. Mater. 2007, 19, 602. 23. Fan, Y; Chen, X.; Trigg, A. D.; Tung, C.-H.; Kong, J; Gao, Z. J.
24. Baba, A.; Tian, S.; Stefani, F.; Xia, C.; Wang, Z.; Advincula, R. C.; Johannsmann, D.; Knoll, W. J. Electroanal. Chem. 2004, 562, 95. 25. Virji, S.; Huang, J.; Kaner, R. B.; Weiller, B. H. Nano Lett. 2004,
4, 491.
26. Schomberg, D.; Salzmann, M.; Stephan, D. Enzyme Handbook; Springer: New York, USA, 1993; Vol. 7, EC 1.11.1.7., p 1. 27. Feast, W. J.; Tsibouklis, J.; Pouwer, K. L.; Groenendaal, L.; Meijer,
E. W. Polymer 1996, 37, 5017.
28. Cruz-Silva, R.; Ruiz-Flores, C.; Arizmendi, L.; Romero-Garcia, J.; Arias-Marin, E.; Moggio, I.; Castillon, F. F.; Farias, M. H.
Poly-mer 2006, 47, 1563.
29. Cruz-Silva, R.; Romero-Garcia, J.; Angulo-Sanchez, J. L.; Le-dezma-Perez, A.; Arias-Marin, E.; Moggio, I.; Farias, M. H. Euro.
Poly. J. 2005, 41, 1129.
30. Sakharov, I. Y.; Alpeeva, I. S.; Efremov, E. E. J. Agric. Food Chem. 2006, 54, 1584.