The Molecular Mechanism of
Trichomonas vaginalis-induced Apoptosis
in Macrophages
Jae-Ho Chang
The Graduate School
Yonsei University
The Molecular Mechanism of
Trichomonas vaginalis-induced Apoptosis
in Macrophages
A Dissertation
Submitted to the Department of Biomedical Laboratory Science
and the Graduate School of Yonsei University
in partial fulfillment of the
requirements for the degree of
This certifies that the dissertation of Jae-Ho Chang is approved.
___________________________
Thesis Supervisor: Yong-Suk Ryang
___________________________ Yong-Serk Park: Thesis Committee Member
___________________________ Hye-Young Lee: Thesis Committee Member
___________________________ Jae-Sook Ryu: Thesis Committee Member
___________________________ In-Hong Choi: Thesis Committee Member
CONTENTS
Abbreviations--- v
List of Figures--- vi
Abstract --- viii
Chapter I. Apoptosis of macrophages induced by Trichomonas vaginalis through the phosphorylation of p38 MAP kinase that locates at downstream of mitochondria- dependent caspase activation--- 1
1. Introduction --- 2
2. Materials and Methods--- 5
Antibodies and other reagents--- 5
Parasites---5
Cell culture and in vitro infection--- 6
Apoptosis analysis--- 6
Western blot analysis---7
Detection of cytochrome c release---7
Transfection---8
Statistical evaluation---8
3. Results---9
T. vaginalis adhesion leaded to sustained p38 activation and rapid ERK activation ---9 T. vaginalis adhesion induced cell death through apoptosis in RAW264.7 cells
T. vaginalis adhesion affected the expression of Bcl-2 family
---13
T. vaginalis-induced apoptosis was mediated by cytochrome c release leading to caspases activation---14
The activation of caspase-3 mediated T. vaginalis-induced apoptosis ---17
T. vaginalis induced apoptosis through the p38 MAPK downstream of caspase 3 pathway---21
4. Discussion---29
Chapter II. Trichomonas vaginalis-induced apoptosis in RAW264.7 cells is regulated through Bcl-xL, but not Bcl-2--- ---33
1. Introduction ---34
2. Materials and Methods---38
Materials---38
Parasites---38
Cell culture and in vitro infection---39
Apoptosis analysis---39
DNA fragmentation analysis---39
Quantitative analysis of apoptosis---40
Western analysis---40
Detection of cytochrome c release---41
expression---43
Bcl-2 overexpression did not protect RAW264.7 cells from the induction of apoptosis by T. vaginalis---47
Bcl-2 overexpression had no influence on the release of cytochrome c, the activation of Bax, caspase-9, -3 and PARP cleavage ---50
T. vaginalis treatment affected expression of Bcl-xL in RAW264.7 cells---54
Transient overexpression of Bcl-xL inhibited T. vaginalis-induced apoptosis in a dose-dependent manner---57
4. Discussion---61
Chapter III. Trichomonas vaginalis inhibits proinflammatory cytokine production in macrophages by suppressing NF-κB activation---65
1. Introduction ---66
2. Materials and Methods---69
Materials---69
Parasites---69
Cell cultures and in vitro infection---69
Transient transfection and luciferase reporter gene assay---70
Electrophoretic mobility shift assay (EMSAs)---70
RT-PCR---71
Quantitative real-time PCR---72
ELISA---73
Quantitative analysis of apoptosis by flow cytometry---73
Western blot analysis---73
Statistical evaluation---74
macrophage cells during early adhesion by T. vaginalis---75
NF-κB activation by T. vaginalis adhesion depended on degradation of IκB-α--78
Prolonged T. vaginalis adhesion inhibited the mRNA expression of TNF-α and IL-12 ---80
T. vaginalis adhesion inhibited TNF-α and IL-12 production accompanied by NF- κB inhibition ---83
Early NF-κB activation in T. vaginalis-adhesive RAW264.7 cells protected apoptotic cell death---87
4. Discussion---89
Conclusions---93
References---95
Abbreviations
Boc-D-FMK : Boc-Asp (OMe)-CH2F
ERK : extracellular signal-regulated kinase
FITC : fluorescein isothiocyanate IL-12 : interleukin-12
JNK : c-jun NH2-terminal kinase
MAPK : mitogen-activated protein kinase
NF-κB : nuclear factor κB
PARP : poly(A)DP-ribose polymerase
PCR : polymerase chain reaction PBS : phosphate-buffered saline
PI : propidium iodide PKC : protein kinase C
RT-PCR : reverse transcription polymerase chain reaction SAPK : stress-activated protein kinase
TNF : tumor necrosis factor
TYM : Trypticasc-yeast extract-maltose
List of Figures
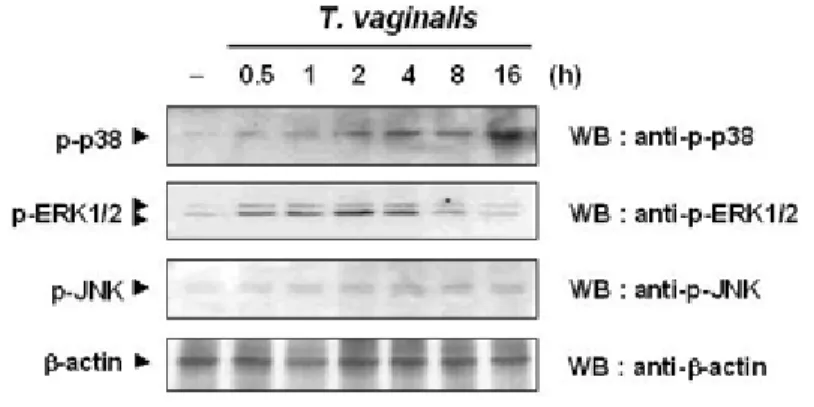
Figure I-1. Time course of MAP kinase activations in response to T. vaginalis---10
Figure I-2. Apoptotic features of T. vaginalis-induced apoptotic cell death---12
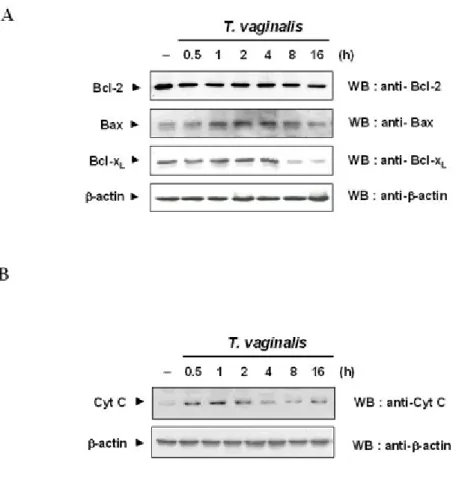
Figure I-3. Immunoblot analysis showing Bcl-2, Bax, Bcl-xL/S and cytochrome c--15
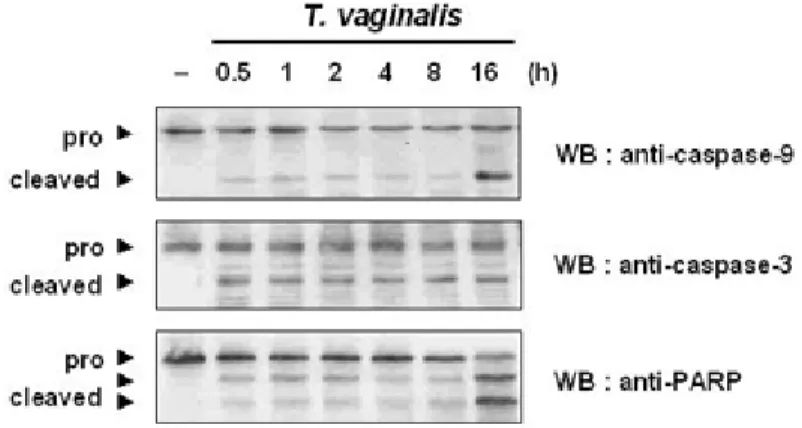
Figure I-4. Effects of caspases on T. vaginalis-induced Apoptosis---16
Figure I-5. Effect of caspase inhibitors on T. vaginalis-induced apoptosis---18
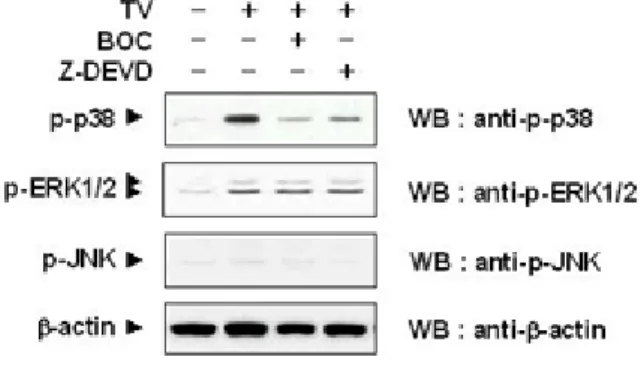
Figure I-6. Effect of caspase inhibitors on activities of p38 MAPK, ERK1/2, and JNK---22
Figure I-7. Effects of p38 MAPK in T. vaginalis-induced apoptosis of RAW 264.7 cells---23
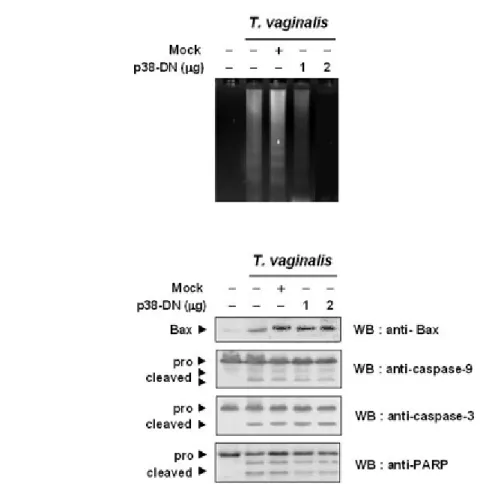
Figure I-8. Effect of dominant negative p38 MAPK on DNA fragmentation and PARP cleavage---26
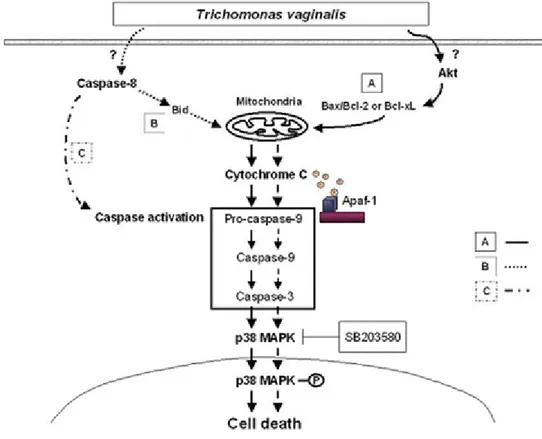
Figure I-9. Putatative pathways of T. vaginalis-induced apoptosis---27
Figure II-1. T. vaginalis-induced apoptosis in RAW264.7 cells---44
Figure II-2. T. vaginalis did not affect Bcl-2 expression in RAW264.7 cells---46
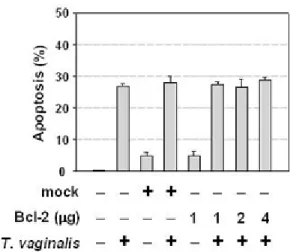
Figure II-3. Overexpression of Bcl-2 did not prevent T. vaginalis-induced apoptosis---48
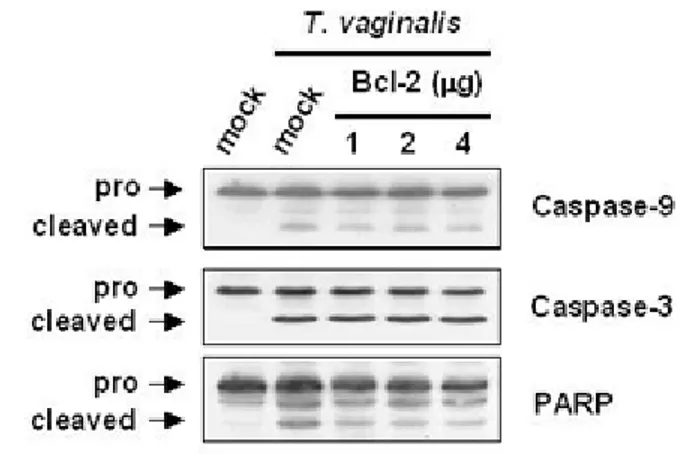
Figure II-4. Overexpression of Bcl-2 had no influence on the release of cytochrome c, the activation of Bax, caspase-9, -3 and PARP---52
Figure III-3. Effect of T. vaginalis adhesion on the mRNA expression of
proinflammatory cytokines TNF-α and IL-12 in macrophages---81
Figure III-4. Effect of T. vaginalis adhesion on the production of proinflammatory cytokines TNF-α and IL-12 in macrophages---84
Figure III-5. Propelled apoptotic cell death by inhibiting NF-κB activity in
ABSTRACT
The Molecular Mechanism of Trichomonas vaginalis-induced Apoptosis in Macrophages
Jae-Ho Chang
Dept. of Biomedical Laboratory Science
The Graduate School Yonsei University
In the present study, the signaling pathways triggered by Trichomonas vaginalis were investigated in
RAW264.7 murine macrophage cell line. T. vaginalis induced mitochondrial changes such as cytochrome c release and the imbalance of the ratio of Bcl-2/Bcl-xL to Bax, and elicited strong phosphorylation of p38
MAPK and negligible ERK1/2 activation. T. vaginalis treatment led to sustained phosphorylation of p38 peaking at 16 h and to transient ERK1/2 phosphorylation that diminished 2 h after incubation. Also, gel
shift analysis showed that NF-κB activation, which was rapidly induced within 1 h, decreased to baseline level with the culture time, then decreased after 16 h post-adhesion. Induction of TNF-α and IL-12 mRNA
was significantly inhibited. These biochemical changes culminated in apoptotic cell death of macrophages. Caspase inhibitors significantly inhibited T. vaginalis-induced apoptotic nuclear damage as well as
activation of p38 MAPK and downregulation of Bcl-xL. Treatment with SB203580, inhibitor of p38
cells treated with T. vaginalis. Additionally, NF-κB binding activity functions an important controlling
factor in cytokine production and survival of T. vaginalis-adhesive RAW264.7 cells. Thus, the present
study suggests the presence of possible modulator mechanisms induced by T. vaginalis to evade host immunity during T. vaginalis infection.
---
Key words: Trichomonas vaginalis, Apoptosis, Monocytes/Macrophages, Signal Transduction, Bcl-2 family, Electrophoretic motility shift assay (EMSA), NF-κB, p38 MAP
CHAPTER I
Apoptosis of macrophages induced by Trichomonas
vaginalis through the phosphorylation of p38 MAP
kinase that locates at downstream of
1. Introduction
Trichomonas vaginalis is a flagellated protozoan parasite, which infects the genito-urinary
tract of humans. It is the causative organism of trichomoniasis, one of the most prevalent sexually transmitted disease (STD) in the world. T. vaginalis infection causes exocervicitis,
vaginitis, and urethritis, and has been also implicated in adverse outcomes of pregnancy, infertility, and increased acquisition of human immunodeficiency virus (Petrin et al., 1998). It
has been reported that adherence to epithelial cells is characteristic of T. vaginalis (Alderete et al., 1995) and that a cytological change observed in trichomoniasis is the detachment of
epithelial cells (Gupta et al., 1990). These findings strongly suggest presence of direct contact of monocytes/macrophages with T. vaginalis in the submucosa.
The innate immune response is the first defense mechanism to protect the host from microbes, limits the infection, and provides signals required for the development of the
adaptive immunity response (Janeway, 2001; Tanya et al. 1997). If not properly regulated by the innate immune system of the host cells, infection by microbes can induce fatal results, whi
ch are already shown in several experimental systems (Freire-de-Lima et al., 1998; Hisaeda
monocytes/macrophges, which plays an important role in the local accumulation of neutrophil to amplify resistance against T. vaginalis infection (Shaio et al., 1995).
Apoptosis of mammalian host cells has been shown to be a hallmark of infection by some bacteria, viruses, and parasites (Payne et al., 1995). Some intracellular pathogens, including
Richettsia rickettsii (Clifton et al., 1998) and Chlamydia trachomatis (Cosulich et al., 1997)
protect infected cells against apoptosis induced by effector cells and soluble mediators of the
immune system, which may allow these organisms to survive intracellularly. Other microbes, such as Yersinia enterocolitica (Ruckdeschel et al., 1997), Shigella flexneri (Zychlinsky et al.,
1992), and Legionella pneumophila (Gao et al., 1999) induce apoptosis of immune effector cells (Clifton et al., 1998). Also, apoptosis of CD4+ T lymphocytes induced by high levels of
NO produced by activated macrophages appears to be an important defense mechanism acting during the acute phase of infection with the opportunistic protozoan parasite Toxoplasma
gondii (Candolfi et al., 1994). Inhibition of apoptosis in T. gondii-infected cells is reported to
be due to profound blockade of caspase activation and caspase activity (Payne et al., 2003).
However, the cellular and molecular mechanisms of pathogenesis by T. vaginalis adhesion, es pecially signaling pathway induced by the infection are not yet well defined.
Induction of apoptosis is associated with a family of cysteine-containing, asparate-specific protease (caspases) (Martin et al., 1995). Two major pathways of apical caspases activation,
Bcl-xL, and A1) as well as pro-apoptotic (Bax and Bak) molecules (Kroemer et al., 1998;
Wolter et al., 1997). Several lines of evidence suggest that ratio of anti- to pro-apoptotic
molecules, such as Bcl-2 or Bcl-xL/Bax, determines the response to a death signal (Ozaki et al.,
1999).
Mitogen-activated protein kinases (MAPKs) are central in many host innate responses triggered by proinflammatory cytokines, products of microorganisms, and environmental
stress (Rouse et al., 1994). The manipulation of host cell MAPK pathways during the infection process may be part of virulence strategies. Many pathogens thus have devised effective
strategies aimed at deactivating or inhibiting MAPK pathways (Priv et al., 2000). MAPKs play a key role in the parasite proliferation, differentiation, and probably in invasion in the
case of T. gondii infection (Robert-Gangneux et al., 2000); however, it remains undetermined w h e t h e r T. vaginalis-induced MAPK activation affects the apoptosis of monocytes
/macrophages and Bcl-2 family gene expression.
In this study, the signaling pathways in macrophage activation by T. vaginalis using
RAW264.7 cells as an infection model were examined. T. vaginalis triggered induction of mitochondrial changes, activation of p38 MAPK, and apoptotic cell death, showing the eviden
2. Materials and Methods
Antibodies and other reagents
Media and serum were purchased from sigma (St. Louis, USA). Anti-ERK1/2, anti-p38, anti-phospho-specific ERK1/2, anti-phospho-p38 MAPK, and anti-PARP were from Santa
Cruz Biotechnology (Santa Cruz, USA). Anti-SAPK/JNK and anti-pospho-specific SAPK/JNK antibodies were from New England Biolabs (Beverly, USA). Boc-D-FMK
(Boc-Asp (OMe)-CH2F and Z-DEVD-FMK (Z-Asp (OMe)-Glu (OMe)-Val-Asp (OMe)-CH2F) were
from Calbiochem (San Diego, USA). Bcl-2, Bcl-xL, and Bax were purchased from Santa Cruz
Biotechnology (Santa Cruz, USA). Cytochrome c was purchased from Calbiochem (San Diego, USA). The monoclonal antibodies to caspase 3 (CPP32) and caspase 9 were from
Transduction Laboratories Inc. Annexin V/PI kit was purchased from Pharmingen (San Diego, USA). SB 203580, a specific inhibitor of p38 MAPK, was obtained from Calbiochem (San
Diego, USA).
Parasites
The Trichomonas vaginalis strain KT-4 (kindly provided by J. S. Ryu, Department of
Parasitology, University of HanYang, Republic of Korea) was used in this study. Trichomonads were cultured in Diamond’s Trypticasc-yeast extract-maltose (TYM) medium
used for assays.
Cell culture and in vitro infection
Murine macrophage derived RAW 264.7 cells were cultured in DMEM (Gibco BRL, Hercules, USA) and supplemented with 10% FBS, penicillin (100 U/ml), and streptomycin (100 U/ml) in an atmosphere of 5% CO2 in air at 37℃. Parasites were treated to RAW264.7
cells (10:1 ratio) and dishes were smoothly rotated to contact.
Apoptosis analysis
Approximately 2 × 106 cells were lysed with 0.2 ml of lysis buffer containing 10 mM
Tris-HCl, 20 mM EDTA, and 0.5 % Triton X-100 (pH 8.0), and placed on ice for 30 min. Cell
extracts were clarified by centrifugation at 14,000 rpm for 10 min. The supernatant containing
DNA cleavage products was precipitated overnight using isopropyl alcohol (Merck, Clarkston, USA). The lysates were incubated with 0.3 mg/ml proteinase K (Boehringer Mannheim,
Mannheim, Germany) at 37℃ for 1 h. Dry DNA pellets were then resuspended in TE buffer (10 mM Tris-HCl, pH 7.5, and 1 mM EDTA), containing 0.5 mg/ml RNase A (Boehringer Mannheim, Mannheim, Germany). DNA fragments were seperated on a 1.8% agarose gel,
(10,000 cells/sample) was analyzed on a FACScan flow cytometer (Becton Dickinson, USA).
Western blot analysis
RAW 264.7 cells were seeded in 35-mm plastic dishes (3 × 105 cells per dish) and incubated
with T. vaginalis for different time periods. Cells were lysed in the lysis buffer (50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 1 µg/ml aprotinin,
10 µg/ml leupeptin, 1 µg/ml pepstatin A, and 1 mM sodium orthovanadate). After centrifugation at 13,000 × g at 4℃ for 30 min, supernatant was collected, 20 µg of lysates
from each sample was run on 10% SDS-polyacrylamide gel and then electrophoretically
transferred to PVDF (polyvinylidene difluoride) membranes. PVDF membranes were rinsed in TBST (10 mM Tris-HCl (pH 7.4), 0.9% NaCl, 0.05% Tween 20, and 1 mM EDTA) and blocked in blocking buffer (TBST containing 5% bovine serum albumin) overnight at 4℃.
PVDF membranes were incubated with primary antibodies overnight at 4℃, washed, and incubated with goat anti-rabbit HRP or anti-mouse HRP for 1 h at room temperature. The me
mbrane was developed with ECL substrate (Amersham Life Sciences, Arlington Heights, USA), and exposed to Biomax MS autoradiography x-ray film (Kodak, Rochester, USA).
Detection of cytochrome c release
homogenized with 30 strokes of a homogenizer (Wheaton, USA). The mitochondria-enriched fraction was pelleted by centrifugation at 14,000 rpm for 30 min. The supernatant was
subjected to Western blot analysis with a monoclonal antibody to cytochrome c.
Transfection
RAW 264.7 cells were transiently transfected with the pcDNA3 vector encoding a
kinase-inactive p38 MAPK, as described previously (Ludwig et al., 1998). Transfection was performed using LipofectAMINE2000 reagent (Life Technologies, USA).
Statistical evaluation
All experiments were performed at least three times. Results are presented as means ± SD.
3. Results
T. vaginalis adhesion leaded to sustained p38 activation, and rapid ERK activation.
First, this study examined time course of p38 MAPK, ERK1/2, and JNK phosphorylation following T. vaginalis adhesion. Figure I-1 shows the levels of phosphorylation of p38 starte
d increasing at 2 h and peaked at 16 h after T. vaginalis adhesion. In contrast, phosphorylation of ERK1/2 increased at 30 min and then gradually diminished at 2 h. A level of JNK
phosphorylation was not affected by T. vaginalis adhesion. Protein expression levels of these kinases were unchanged by T. vaginalis adhesion.
Figure I-1. Time course of MAP kinase activations in response to T. vaginalis. Cells were
treated with T. vaginalis for 0.5-16 h. Lysates were subjected to SDS-PAGE and immunoblotted with anti-phospho-p38, anti-phospho-ERK1/2, and anti-phospho-SAPK/JNK
T. vaginalis adhesion induced cell death through apoptosis in RAW264.7 cells.
To test whether T. vaginalis induces apoptosis in RAW264.7 cells, cell death was monitored
by Annexin-V staining and by DNA fragmentation. FACS analysis revealed that T. vaginalis adhesion significantly increased the number of apoptotic cells (P < 0.05) (Figure I-2A). I
ncrease in apoptotic cells was also confirmed by measuring the formation of hypodiploid DNA. Electrophoretic patterns of laddered oligonucleosomal DNA fragments were observed
in T. vaginalis-adhesive RAW264.7 cells (Figure I-2B).
Figure I-2. Apoptotic features of T. vaginalis-induced apoptotic cell death. RAW264.7 cells
were cultured with (T.V.) or without T. vaginalis (none) and then cells were harvested at 8 h after T. vaginalis adhesion. (A) For FACS analysis, cells were stained with FITC-conjugated
T. vaginalis adhesion affected the expression of Bcl-2 family.
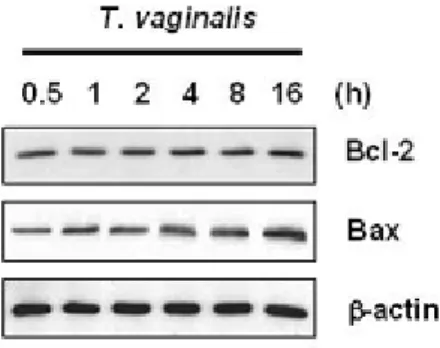
To investigate whether the ratio of Bcl-2 or Bcl-xL to Bax is associated with T.
vaginalis-induced apoptosis, RAW264.7 cells were incubated with T. vaginalis for indicated time peri od, and then the lysates were blotted with anti-Bcl-2, anti-Bcl-xL, and anti-Bax antibodies. D
egradation of Bcl-xL and a band corresponding to Bcl-xs was observed after 2 h
post-adhesion as depicted in Figure I-3A. Bcl-2 protein in RAW264.7 cells remained unchanged
throughout the course of apoptotic death in response to T. vaginalis adhesion, while the expression of Bax protein was slightly increased during entire course of culture.
T. vaginalis-induced apoptosis was mediated by cytochrome c release leading to caspases
activation.
It is known well that Bcl-2 and Bcl-xL prevents apoptosis by inhibiting cytochrome c release, while Bax stimulates cytochrome c release from mitochondria. This study first asked
whether cytochrome c release is involved in T. vaginalis-induced apoptosis by isolating cytosolic fraction from cultured RAW264.7 cells treated with T. vaginalis and by checking the
presence of cytochrome c by Western blot with specific antibody. Figure I-3B demonstrates that cytochrome c release started as early as 0.5 h post infection and peaked at 1 h.
Then the expression of caspase-9, caspase-3, and PARP in order to know sequence of e vents that link mitochondrial cytochrome c to other molecules was investigated. Figure I-4
demonstrates that caspase-9 is cleaved to p37 and p35 fragments at 0.5 h of infection. The cle avage of caspase-9 peaked at 16 h post infection. Caspase-3 is cleaved from the proenzyme
(32-kDa) to active form (18-kDa) in T. vaginalis-induced apoptosis at 0.5 h post incubation, and the amount of cleaved products stayed at the same level throughout the culture. T.
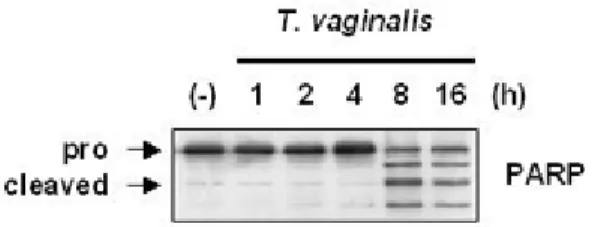
vaginalis adhesion induced the cleavage of 116-kDa PARP into the fragment COOH-terminal
85-kDa in RAW264.7 cells at 0.5 h post-adhesion, and again the cleavage peaked at 16 h.
Figure I-3. Immunoblot analysis showing Bcl-2, Bax, Bcl-xL/S and cytochrome c. (A) RAW
264.7 cells were treated with T. vaginalis for different times, and blotted with Bcl-2, anti-BclxL, and anti Bax antibodies as indicated. Results are representative of three separate
experiments with comparable outcomes. (B) Time course release of cytochrome c in RAW 264.7 cells. RAW 264.7 cells were treated to T. vaginalis for 0.5-16 h. Cells were collected at
Figure I-4. Effects of caspases on T. vaginalis-induced apoptosis. After RAW 264.7 cells were
treated with T. vaginalis for indicated times, imminoblot analyses for caspase-9, caspase-3, and PARP.
The activation of caspase-3 mediated T. vaginalis-induced apoptosis.
To clarify the role of caspase activation on T. vaginalis-induced apoptosis, Z-DEVD-FMK
and Boc-D-FMK, which are inhibitors of caspase-3 and broad-caspases, respectively, were added to the culture medium 30 min prior to exposure to T. vaginalis. As expected, both
caspase inhibitors significantly suppressed T. vaginalis-induced apoptosis and DNA fragmentation (Figure I-5A, B). In addition, the cleavage of caspase-3 and the cleavage of
PARP were completely blocked in the presence of the broad-caspases inhibitor and caspase-3 inhibitor (Figure I-5C). However, the addition of caspase inhibitors did not prevent the release
of cytochrome c (Figure I-5D). These findings again support the idea that cytochrome c rele ase precedes activation of caspases.
Figure I-5. Effect of caspase inhibitors on T. vaginalis-induced apoptosis. After RAW 264.7 cells were pre-incubated with or without 50 µM Boc-D-FMK and 50 µM Z-DEVD-FMK for
30 min, cells were treated with or without T. vaginalis for 8 h. (A) Cells were stained by FITC-conjugated annexin V and propidium iodide (PI) and then analyzed on flow cytometry.
Results are expressed as means ±SD and are representative of three independent experiments. (B) The formation of oligonucleosomal fragments was determined by agarose gel
electrophoresis. Similar results were achieved in three separate experiments with comparable outcomes. (C) Cells were treated with T. vaginalis for indicated times and then immunoblot analysis was followed to detect caspase-9, caspase-3, and PARP cleavage. Anti-β-actin
antibody was used to confirm equal loading of the extracts. (D) RAW 264.7 cells were treated to T. vaginalis in the absence (-) or presence (+) of 50 µM Boc-D-FMK and 50 µM
Z-DEVD-FMK. Caspase inhibitors were added to the culture medium 30 min prior to the
treatment of T. vaginalis. Cells were harvested at 8 h after the T. vaginalis adhesion and cytosolic fractions were subjected to immunoblot analysis with a monoclonal antibody to
T. vaginalis induced apoptosis through the p38 MAPK downstream of caspase 3 pathway.
p38 MAPK was rapidly activated and sustained in RAW264.7 cells during entire course of
culture with T. vaginalis, whrease only minimal activation of JNK is detected (Figure I-1). To identify further detailed pathways involved in T. vaginalis-induced apoptosis, it was
determined the cross-talk between p38 MAPK and caspase proteases using Z-DEVD-FMK, a known caspase-3 inhibitor, and Boc-D-FMK, a broad caspase inhibitor. Both caspase
inhibitors blocked the activation of p38 MAPK, indicating that p38 MAPK functioned at dow nstream of caspase activation. The activation of ERK1/2 and JNK did not affected by those i nhibitors (Figure I-6). Moreover, treatment with 10 µΜ SB203580, a MAPK inhibitor,
protected cell death induced by T. vaginalis, which was assessed by FACS analysis (Figure
I-7A) and DNA fragmentation assay (Figure I-7B). In contrast, SB203580 did not block Bax activation, cytochrome c release from mitochondria (Figure I-7C), caspase-9, -3 activation, or
PARP cleavage at 16 h after T. vaginalis treatment (Figure I-7D). More importantly, the overexpression of kinase-inactive p38 MAPK did not prevent T. vaginalis-induced Bax,
caspase-9, -3, and PARP cleavage although it led to attenuated cell death (Figure I-8A, B). These data strongly suggest that the cleavage of caspase-9 and caspase-3 is required for
activation of p38 MAPK and that phosphorylation of p38 MAPK is located downstream of caspase pathway.
Figure I-6. Effect of caspase inhibitors on activities of p38 MAPK, ERK1/2, and JNK. 50 µM Boc-D-FMK and 50 µM Z-DEVD-FMK were added to the culture medium for 30 min prior to
the treatment of T. vaginalis. After treatment with T. vaginalis for 8 h, RAW 264.7 cells were subjected to immunoblotting with phospho-ERK, phospho-SAPK/JNK, and
anti-phospho-p38 MAPK antibodies. The results are from a representative study performed three times with comparable outcomes.
Figure I-7. Effects of p38 MAPK in T. vaginalis-induced apoptosis of RAW 264.7 cells. After RAW 264.7 cells were pre-incubated with or without p38 MAPK inhibitor SB203580 (5 µM and 10 µM) for 30 min, cells were treated with or without T. vaginalis for 8 h. (A) Apoptosis
of RAW 264.7 cells was assessed with annexin V-FITC and PI assay. Results are the means of
three independent experiments. (B) The formation of oligonucleosomal fragments was determined by agarose gel electrophoresis. Similar results were achieved in three separate
experiments with comparable outcomes. (C) RAW 264.7 cells were treated with T. vaginalis for different times, and blotted with anti-Bax antibody as indicated. Cytosolic fractions were
subjected to western blotting analysis with a monoclonal antibody to cytochrome c. (D) Cells were pre-treated with 5 µM and 10 µM SB203580 for 30 min and then T. vaginalis was treated for 8 h. Immunoblot analyses for caspase-9, -3 and PARP were performed. Anti-β-actin
Figure I-8. Effect of dominant negative p38 MAPK on DNA fragmentation and PARP cleavage. RAW 264.7 cells were transiently transfected with the pcDNA3 vector encoding a
Figure I-9. Putatative pathways of T. vaginalis-induced apoptosis. Pathway A deserves specific
attention because it is activated in the RAW264.7 cells expressing Bcl-2 family, presumably by inhibiting Akt activation. In this pathway, T. vaginalis increases Bax protein with reducing
Pathway B represents that caspase-8 probably may act indirectly leading to the release of cytochrome c and activation of caspase-9 with subsequent activation of the effector caspases.
The Bid-mediated cytochrome c release and procapase-9 activation seems to play a minor role in T. vaginalis-induced apoptosis. Pathway C may play a prominent role in
caspase-8-expressing cells. In this case, procaspase 8 is activated by unknown mechanisms. Active caspase-8 then activates the effector caspase-3-like proteases, but this pathway does not seem
4. Discussion
Induction of apoptosis of mammalian host cells has been shown for several parasites
including T. gondii, L. donovani, and E. histolytica (Kasper et al., 200; Priv et al., 2000). However, the signaling machinery for cell death of host cells triggered by T. vaginalis is still
largely unknown.
The present study demonstrates the induction of apoptosis in murine macrophages upon
adhesion with T. vaginalis and provides insight into the molecular details triggering apoptosis through p38 activation. The exact functional role of p38 MAPK in cellular stress response
remains less well understood and controversial. Some studies have observed that p38 MAPK activation is dependent on the initiator caspase activation and, in some cases, unrelated to the
apoptosis (Mackey et al., 1997; Ozaki et al., 1999; Zhang et al., 2000). Other study has shown that p38 MAPK activation occurs either upstream or independent of caspases and mediates
apoptosis (Frasch et al., 1998). However, the involvement of p38 MAPK in apoptosis, especially in parasites, is ambiguous so far.
This study provides the evidence that the sustained activity of p38 MAPK is required for the induction of apoptotic cell death by T. vaginalis in RAW264.7 cells. It is in concordance with
the data observed in other parasitic protozoa, such as T. gondii and L. donovani (Junghae et
between ERK and p38 MAPK after T. vaginalis adhesion may be an important factor in the evasion from macrophages and in innate immune responses.
Recent studies demonstrate that Bcl-2 family plays a central role in decision of cell survival and cell death at the level of mitochondria, which is controlled by the amounts of Bcl-2-Bax
or Bcl-xL-Bax heterodimers (Adams et al., 1998; Reed, 1994). In this study, Bcl-xL expression
was decreased after T. vaginalis adhesion, whereas the expression of Bcl-2 protein remained
unchanged. Bcl-2 and Bcl-xL are capable of blocking cytochrome c release in cell-free extracts
and in cells treated with apoptosis-inducing agents (Kluck et al., 1997; Reed, 1994). Therefore,
imbalance of the ratio of Bcl-2 or Bcl-xL to Bax may play a pivotal role in T.
vaginalis-induced apoptosis by regulating mitochondrial cytochrome c release.
The present study has shown in this paper that caspases are involved in the apoptosis observed during T. vaginalis adhesion. Thesedata are in agreement with published data
showing that the protozoan parasite C. parvum leads to apoptotic nuclear condensation and DNA fragmentation in host cells, and apoptosis was inhibited by caspase inhibitors (Ojcius et
al., 1999). Furthermore, it was demonstrated that the cytochrome c release from mitochondria occurs independently of caspase activation and of phosphorylation of p38 MAPK that is in
Activation of caspase-8 was not detected throughout the course of T. vaginalis-induced apoptosis, although T. vaginalis-induced apoptosis was effectively suppressed in the presence
of a broad caspase inhibitor, Boc-D-FMK. These results demonstrate that caspase-8 is dispensable for T. vaginalis-induced activation of caspase-3 and apoptosis at least in
RAW264.7 cells.
T. vaginalis induce apoptotic cell death through perturbation of apoptotic signal
transduction pathways in mitochondria during infection process. Why does T. vaginalis induce apoptosis in infected cells? It appears to us that T. vaginalis avoids its own death by
modulating the effector cells by blocking fusion of the protozoa to endocytic vesicles that allow phagosomal maturation (Cosulich et al., 1997; Freire-de-Lima et al., 1998). Death of m
acrophage would also reduce inflammation at infection site. Control of local inflammation and local immune response may be beneficial for host in some way to maintain stable ho
st-parasite relationship without causing severe inflammation and/or tissue damage. In any event, the induction of apoptosis seems to be initial step in the specific immune response to T.
vaginalis. It would require more detailed analysis in order to address this question.
This study now has picture showing signaling pathway induced by T. vaginalis infecti
on in macrophage and how this infection leads to cell death (Figure I-9). Bcl-2 family is involved in the caspase-dependent pathway of T. vaginalis-induced apoptosis, resulting in
CHAPTER II
Trichomonas vaginalis-induced apoptosis in RAW264.7
1. Introduction
Trichomonas vaginalis is a protozoan parasite, which infects the genito-urinary tract of
humans. It is responsible for trichomoniasis, one of the most common nonviral sexually transmitted disease (STD) in the world. In women, T. vaginalis infection has been associated
with adverse outcomes of pregnancy, infertility, low-birth-weight infants, cervical cancer, and increase in the transmission of human immunodeficiency virus (Cotch et al., 1997; Petrin et
al., 1998). It is reported that adherence to epithelial cell is characteristic of T. vaginalis and
that detachment of epithelial cells by T. vaginalis could result in direct contact of
monocyte/macrophages in the submucosa with T. vaginalis (Alderete et al., 1985; Alderete et
al., 1995).
The induced innate immune responses either succeed in clearing the infection or contain it while an adaptive response develops. Adaptive immunity, greater precisely, harnesses many of
the same effector mechanisms that are used in the innate immune system (Moore et al., 1993; Tanya et al., 1997). Then, antigen-specific T cells activate the microbicidal and
cytokine-secreting properties of macrophages harboring pathogen (Abrahamsohn et al., 1996). Macrophages are important effectors of the innate immune system, and are capable of
al., 2002). Recent report has shown that IL-8 production by triggered T. vaginalis in
monocytes/macrophages, particularly adherent trophozoites, plays an important role in the
local accumulation of neutrophil to increase resistance against T. vaginalis infection (Shaio et
al., 1995). In a murine model of experimental trichomoniasis, specific IgA antibodies might
help to protect asymptomatic individuals from severe infection and T-lymphocytes may play an important function in the eradication of the parasite (Paintlia et al., 2002). The study of
immunomodulator activity of natural or synthetic drugs proves usefulness of the murine model of experimental trichomoniasis (Nogal-Ruiz et al., 2003). However, the precise cell biological
mechanisms of immune response in T. vaginalis infection remained unknown.
Apoptosis is a highly regulated mechanism of maintaining homeostasis of multicellular
organisms and in modulating pathogenesis of a variety of disease (Usher et al., 2002). T.
gondii and Trypanosoma cruzi inhibits apoptosis by blocking blockade of caspase activity
(Payne et al., 2003; Nakajima-Shimada et al., 2000). Other obligate intracellualr pathogens, such as Rickettsia rickettsii and Brucella melitensis, inhibit host cell apoptosis, which may
benefit these organisms to grow and survive intracellularly (Clifton et al., 1998; Fernandez-Prada et al., 2003). On the other hand, in host cells via interleukin-12- and
Fas/FasL-dependent pathway, T. gondii induce apoptosis (Gavrilescu et al., 2003). Also, other parasites, such as Legionella pneumophila, and Entamoeba histolytica, induce apoptosis in macrophages,
Mitochondria have been shown to be a target for mediators of apoptosis and play a central role in the apoptosis in many cell types. Especially, Bcl-2, and Bcl-xL, two members of Bcl-2
family, play central roles in the regulation of apoptosis and are considered homologs of the nematode Caenorhabditis elegans CED-9 (Kroemer et al., 1998; Chao et al., 1997). Genetic
studies from C. elegans have identified several key regulators that play important roles in the induction and execution of apoptosis: CED-3, CED-4, and CED-9 (Ellis et al., 1986). Two of
the genes, ced-3 and ced-4, are necessary for execution of apoptosis. Bcl-2 and Bcl-xL have
been shown to be capable of blocking the release of cytochrome c, therby preventing the
activation of caspase-9 and its downstream caspases. In contrast, Bax has been shown to be a pro-apoptotic Bcl-2 family protein, which triggers the release of cytochrome c from
mitochodria (Oltvai et al., 1993; Wolter et al., 1997). Bcl-2 is found in several intracellular membranes, including the outer mitochondrial membrane, the endoplasmic reticulum (ER),
and the nuclear envelope (Farrow et al., 1995). However, Bax is not located in ER but remains in the cytosol until an apoptotic signal is generated and then induces its translocation to
mitochondria (Akao et al., 1994). In contrast, several studies also revealed that 2 and Bcl-xL under certain conditions could function as pro-apoptotic molecules. Bcl-2 and Bcl-xL can be
In the present study, I examined the role of Bcl-2 family in the control of T. vaginalis-inducedapoptosis in RAW 264.7 cells. Bcl-2 overexpression has little influence on T.
vaginalis-induced apoptotic cell death. In contrast, T. vaginalis-induced apoptotic cell death
was prevented in RAW 264.7 cells by transfection with Bcl-xL. Furthermore, RAW 264.7 cells
overexpressing Bcl-xL are resistant to T. vaginalis-induced cytochrome c release and caspase-9
activation. This finding may reflect that the anti-apoptotic effect of Bcl-xL, not Bcl-2, occurs
2. Materials and Methods
Materials
Media and serum were purchased from Sigma (Gibco BRL, USA). Anti-Bcl-2, -Bcl-xL, and
-Bax antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, USA).
Boc-D-FMK (Boc-Asp (OMe)-CH2F), Z-DEVD-FMK
(Z-Asp(OMe)-Glu(OMe)-Val-Asp(OMe)-CH2F), and Cytochrome c was purchased from Calbiochem (San Diego, USA). The
monoclonal antibodies to caspase-3 (CPP32) and caspase-9 were from Transduction Laboratories Inc. Annexin V/PI kit was purchased from Pharmingen (San Diego, USA).
Chloromethyl-X-rosamine (MitoTracker Red) was from Molecular Probes (Eugene, USA). All other chemicals were analytical-grade reagents.
Parasites
The Trichomonas vaginalis strain KT-4 (kindly provided by J. S. Ryu, Department of Parasitology, University of HanYang, Republic of Korea) was used in this study.
Trichomonads were cultured in Diamond’s Trypticasc-yeast extract-maltose (TYM) medium (Diamond, 1957) and supplemented with 10% heat-inactivated horse serum in an atmosphere
Cell lines culture and in vitro infection
The murine monocyte/macrophage cell line, RAW 264.7, was cultured in DMEM and supplemented with 10% FBS, penicillin (100 U/ml), and streptomycin (100 U/ml) in a humidified atmosphere of 95% air and 5% CO2 at 37℃. Parasites were treated to RAW264.7
cells (10:1 ratio) and dishes were smoothly rotated to contact. At time indicated, supernatants were removed and fresh medium was added.
Apoptosis assay
RAW 264.7 cells were seeded in 35-mm plastic dishes (3 × 105 cells per dish) and treated
with T. vaginalis at a parasite/cell ratio of 10:1 for 8 h or with T. vaginalis and APC
(pretreatment for 16 h). Cells were prepared and analyzed using APOPercentageTM apoptosis assay according to the manufacturer’s instructions (Biocolor Ltd., USA).
DNA fragmentation analysis
Approximately 2 × 106 cells were lysed with 0.2 ml of lysis buffer containing 10 mM
Tris-HCl, 20 mM EDTA, and 0.5 % Triton X-100 (pH 8.0), and placed on ice for 30 min. Cell
extracts were clarified by centrifugation at 14,000 rpm for 10 min. The supernatant containing DNA cleavage products was precipitated overnight using isopropyl alcohol (Merck, Clarkston,
and visualized by ethidium bromide staining (Bio-Rad, Hercules, USA), and photographed.
Quantitative analysis of apoptosis by flow cytometry
Cells were harvested, washed with PBS, and resuspended in a binding buffer (10 mM Hepes,
pH 7.4, 140 mM NaCl, 2.5 mM CaCl2). After 15 min of incubation with annexin V-fluorescein
isothiocyanate (Sigma, St. Louis, USA) and propidium iodide (Pharmingen, San Diego, USA)
at room temperature, the fluorescence emitted by cells (10,000 cells/sample) was analyzed on a FACScan flow cytometer (Becton-Dickinson, USA).
Western blot analysis
RAW 264.7 cells were seeded in 35-mm plastic dishes (3 × 105 cells per dish) and incubated
with T. vaginalis for different time periods. Cells were lysed in the lysis buffer (50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 1 µg/ml aprotinin,
10 µg/ml leupeptin, 1 µg/ml pepstatin A, and 1 mM sodium orthovanadate). After centrifugation at 13,000 × g at 4℃ for 30 min, 20 µg of supernatant lysates from each sample
was run on 10% SDS-polyacrylamide gel and then electrophoretically transferred to PVDF
Tris-(Amersham Life Sciences, Arlington Heights, USA), and exposed to Biomax MS autoradiography x-ray film (Kodak, Rochester, USA).
Detection of cytochrome c release
1 × 107 cells were trypsinized and collected at the indicated times. Pellets were washed with
ice-cold phosphate buffered saline and resuspended in 125 µl of cytosol extraction buffer (250
mM sucrose, 20 mM Hepes-KOH, pH 7.5, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM
EGTA, 1 mM Dithiothreitol, and 0.1 mM phenylmethylsulfonyl fluoride). The pellets were
then homogenized with 30 strokes of a homogenizer (Wheaton, USA). After the centrifugation at 14,000 rpm for 30 min, the supernatant was collected and used to detect cytochrome c by
Western blotting.
Mitochondrial energization
Mitochondrial energization was determined as the retention of the fluorescent dye MitoTracker Red CMXRos. After T. vaginalis treatment, 1 × 106 cells in 2 ml of complete
DMEM were loaded with the probe MitoTracker Red (1 nM) for 15 min at 37℃before the
flow cytometric analysis. The same incubation time was applied to the control and the T.
vaginalis-treated samples. MitoTracker Red was excited at 579 nm, and fluorescence was
Transfection was performed using LipofectAMINE2000 and PLUS reagent (Invitrogen,
CA), according to the supplier’s protocol. RAW 264.7 cells were plated in 6-well plates (1 ×
106 cells/well) and transfected with 1, 2, and 4 µg of each expression vector (pcDNA3-Bcl-2, or pcDNA3-Bcl-xL-Flag) or with a control vector (pcDNA3, empty vector). The plasmids used
in this experiment were kindly provided by Dr. Lee (University of Yonsei, Republic of Korea). Twenty-four hours after transfection, the cells were treated with T. vaginalis for different time
periods as indicated for western blot analysis and quantitative analysis of apoptosis by flow cytometry.
Statistical evaluation
All experiments were performed at least three times. Results are presented as means ±
standard deviation (SD) if not otherwise indicated. Significance of the results was analyzed by
3. Results
T. vaginalis-induced apoptosis in RAW264.7 cells was independent of Bcl-2 expression.
I hypothesized that target cell death is due to the triggering of apoptosis by T. vaginalis and tested whether T. vaginalis induced apoptosis in RAW264.7 cells. After 8 h incubation with T.
vaginalis, approximately 26% of T. vaginalis-treated RAW264.7 cells were apoptotic, and
subsequently most of them were detached from the tissue culture dish (Figure II-1A, B). It has
been previously demonstrated that caspase-3 is involved in the execution of T. vaginalis-induced apoptosis in RAW264.7 cells (data not shown). I tested that the involvement of PARP
(poly ADP-ribose polymerase) cleavage, which has been correlated with caspase-3 activity, by immunoblotting with anti-PARP antibody. During apoptotic cell death, cleavage of the
116-kDa PARP into its 85-116-kDa fragment was prominent after 8 h in T. vaginalis-treated RAW264.7 cells (Figure II-1C).
Next I investigated the involvement of Bcl-2 family in RAW264.7 cells after T. vaginalis treatment. Western blot analysis revealed that the Bcl-2 protein in RAW264.7 cells was
unchanged throughout the course of apoptotic death after T. vaginalis treatment, while the expression of Bax protein was slightly increased with the culture time (Figure II-2).
Figure II-1. T. vaginalis-induced apoptosis in RAW264.7 cells. (A) T. vaginalis were
incubated with RAW264.7 cells in a 10:1 ratio for 8 h to induce apoptosis. After the T.
vaginalis adhesion, cells undergoing apoptosis were detected by the uptake of a purple dye
(APOPercentage Apoptotic Assay, Biocolor). (B) Flow cytometric analysis of apoptotic cells was performed using annexin V-FITC. Cells were incubated with annexin V-FITC in a buffer
containing iodide (PI) and were analyzed by flow cytometry. Annexin V-FITC and PI positive cells were considered as the apoptotic cells. (C) Cleavage of PARP was detected by immunoblotting anti-PARP antibody in T. vaginalis-treated RAW264.7 cells.
Figure II-2. T. vaginalis did not affect Bcl-2 expression in RAW264.7 cells. RAW 264.7 cells were treated with T. vaginalis for the indicated times and lysed, and equal amounts of protein
were separated using SDS-polyacrylamide gel electrophoresis and probed by western blot with anti-Bcl-2 and anti-Bax antibodies. Results are representative of three separate experiments
Bcl-2 overexpression did not protect RAW264.7 cells from the induction of apoptosis by T.
vaginalis.
To further examine the correlation between Bcl-2 expression and T. vaginalis-induced apoptosis, we tested the ability of Bcl-2 expression to prevent apoptosis by T. vaginalis in
RAW264.7 cells. RAW264.7 cells were transiently transfected with Bcl-2 DNA prior to T.
vaginalis treatment. Immunoblotting confirmed that the transfectants expressed high levels of
Bcl-2 (Figure II-3A). To assess whether Bcl-2 overexpression affects DNA fragmentation in RAW264.7 cells, we performed a DNA fragmentation assay as described in “Materials and
methods.” Bcl-2 overexpression did not attenuate DNA fragmentation (Figure II-3B). Moreover, the quantitative analysis of these effects showed that the apoptotic cell death
induced T. vaginalis can be independent of Bcl-2 (Figure II-3C), suggesting that T. vaginalis-induced apoptosis is indeed refractory to the Bcl-2 overexpresssion.
Figure II-3. Overexpression of Bcl-2 did not prevent T. vaginalis-induced apoptosis. (A)
Overexpressed Bcl-2 expression was determined by immunoblotting in a dose-dependent manner. Immunoblotting with anti-β-actin antibody was used as a loading control. (B) RAW
264.7 cells were transiently transfected with an empty vector as a control or pcDNA3/Bcl-2 and analyzed for DNA fragmentation at specific time points post-transfection. (C) Cells were
stained with Annexin V-FITC and analyzed by flow cytometry, and then double positive cells were considered as the apoptotic cells. Results are expressed as means the means ±SD and
Bcl-2 overexpression had no influence on the release of cytochrome c, the activation of Bax, caspase-9, -3 and PARP cleavage.
Recent studies demonstrated that the release of cytochrome c from the mitochondria to the cytosol is an early step in the apoptotic pathway (Bossy-Wetzel et al., 1998; Kluck et al.,
1997). Previous my study demonstrated that release of cytochrome c started as early as 0.5 h post infection and peaked at 1 h. In this system, I examined whether cytochrome c release was
also inhibited by the Bcl-2 overexpression. The Bcl-2 overexpression had no effect on release of cytochrome c and Bax activity in T. vaginalis-treated RAW264.7 cells, indicating that this
event was not blocked by Bcl-2 expression (Figure II-4A). Furthermore, when I measured mitochondrial transmembrane potential by incubating samples with a MitoTracker probe, the
membrane potential was significantly decreased at 8 h. At this time, only 7% of T. vaginalis-treated RAW264.7 cells were MitoTracker probe positive compared to 45% in unvaginalis-treated cells.
However, the overexpression of Bcl-2 did not affect the change of the membrane potential by
T. vaginalis (Figure II-4B). These results suggest that T. vaginalis-induced cytochrome c
release and Bax activation did not under the inhibitory control of Bcl-2.
Previously data from my study showed that T. vaginalis induced apoptosis through a
Figure II-4. Overexpression of Bcl-2 had no influence on the release of cytochrome c, the
activation of Bax, caspase-9, -3 and PARP cleavage. RAW 264.7 cells were transiently transfected with an empty vector or pcDNA3/Bcl-2. Then, T. vaginalis was added to
RAW264.7 cells for 8 h. (A) Western blot was conducted with the cytochrome c and anti-Bax antibodies as indicated. Overexpressed Bcl-2 expression was determined by
immunoblotting in a dose-dependent manner. (B) Significant change of the membrane potential was observed in RAW 264.7 cells overexpressing Bcl-2. 8 h after T. vaginalis
treatment, cells were collected and stained with MitoTracker dye to measure mitochondrial membrane potential. FACS analysis showed that 18-19% of cells overexpressing Bcl-2
exhibited intense red fluoresence compared to cells transfected with control expresssion vector (50%). FL3-H, red fluorescence intensity; M1, quantification marker of strongly fluorescent cells. (C) Cleavage of caspase-9, caspase-3, and PARP was detected by immunoblotting with
T. vaginalis treatment affected expression of Bcl-xL in RAW264.7 cells.
Anti-apoptotic protein Bcl-xL also has been reported to play critical roles in inhibiting the
release of cytochrome c and subsequent apoptosis (Pan et al., 1998). To test this, I examined the correlation between Bcl-xL expression and T. vaginalis-induced apoptosis. Whereas
untreated RAW264.7 cells showed high expression of Bcl-xL, Bcl-xL expression in T.
vaginalis-treated cells was decreased after 8 h of culture time (Figure II-5A). These data
suggest that alteration of the Bax/Bcl-xL ratio by downregulating Bcl-xL and stabilizing Bax
expression may be critical for induction of apoptosis by T. vaginalis.
Previous work has shown that caspase-3 is involved in the regulation of Bcl-2 expression (Clem et al., 1998). To test this correlation between caspase-3 and Bcl-2 family, I examined T.
vaginalis-treated RAW264.7 cells in the presence or absence of caspase inhibitors
(Boc-D-FMK and Z-DEVD-(Boc-D-FMK). Subsequent western blot analysis of Bcl-xL expression showed that
T. vaginalis-induced downregulation of Bcl-xL was significantly abolished in the presence of
caspase inhibitors. These data, the cleavage of Bcl-xL can be blocked by the addition of the
caspase-3 inhibitors, suggest that the cleavage of Bcl-xL in response to T. vaginalis may be a
Figure II-5. T. vaginalis treatment affected Bcl-xL expression in RAW264.7 cells. (A) T.
vaginalis was added to RAW264.7 cells for the indicated times and lysed, and whole cell
lysate was electrophoresed and probed by western blot with anti-Bcl-xL antibody. (B) 50 µM
Boc-D-FMK, and 50 µM Z-DEVD-FMK were pretreated for 30 min prior to T. vaginalis
adhesion. After T. vagianlis treatment for 8 h, RAW 264.7 cells were subjected to immunoblotting with anti-Bcl-2, anti-Bcl-xL, and anti-Bax antibodies. Results are
representative of three separate experiments with comparable outcomes. Actin was used as loading control. Immunoblotting with anti-β-actin antibody was used as a loading control.
Transient overexpression of Bcl-xL inhibited T. vaginalis-induced apoptosis in a
dose-dependent manner.
My observation that T. vaginalis down regulates Bcl-xL protein suggests that loss of Bcl-xL
is essential for T. vaginalis-induced apoptosis in RAW 264.7 cells. If so, prevention of this loss
would be expected to protect cells from T. vaginalis-induced apoptosis. To confirm the result above, RAW 264.7 cells were transiently transfected with Bcl-xL-Flag and then treated with T.
vaginalis for 8 h. Following treatment, apoptotic cells were quantified. T. vaginalis-induced
apoptosis was dramatically prevented in RAW 264.7 cells by transfection with Bcl-xL-Flag,
whereas a control vector (pcDNA3) had no effect on the ability of T. vaginalis to induce apoptosis (Figure II-6B). These data suggest that Bcl-xL may regulate apoptosis induced by T.
vaginalis in RAW 264.7 cells.
Because I have observed that T. vaginalis induces cytochrome c release and activates
caspase-9, We examined whether Bcl-xL overexpression could prevent these T.
vaginalis-regulated effects. Western blot analysis revealed that Bcl-xL overexpression inhibited
Figure II-6. Overexpression of Bcl-xL blocked T. vaginalis-induced apoptosis. RAW 264.7
cells were transiently transfected with an empty vector (pcDNA3) or Bcl-xL-Flag. Then, T.
vaginalis was added to RAW264.7 cells for 8 h. (A) Overexpressed Bcl-xL expression was
determined by immunoblotting using anti-Flag antibody. (B) Cells were stained with Annexin
V-FITC and analyzed by flow cytometry. (C) Western blot was conducted with the anti-cytochrome c antibody as indicated. Overexpressed Bcl-xL expression was determined by
immunoblotting using anti-Flag antibody. (D) Cleavage of caspase-9 and PARP was detected by immunoblotting with corresponding antibody. The results are representative of three
separate experiments with comparable outcomes. Immunoblotting with anti-β-actin antibody was used as a loading control.
4. Discussion
It is well known that intracellular protozoan parasites such as T. cruzi and T. gondii inhibit
host cell apoptosis in infected cells. Those studies reported that no significant change of the amount of intracellular Bcl-2 was observed in infected cells (Clark et al., 1999; Goebel et al.,
2001). In contrast, E. histolytica induce apoptosis in target cells, which is not blocked by Bcl-2 (Ragland et al., 1994).
In this study, to evaluate the relationship between T. vaginalis infection and cell death, we investigated the roles of Bcl-2 family on induction of apoptosis in T. vaginalis-treated RAW
264.7 cells. The expression of Bcl-2 protein remained almost constant upon treatment with T.
vaginalis (Figure II-2), and overexpression of Bcl-2 did not block T. vaginalis-induced
apoptosis in RAW264.7 cells (Figure II-3). Other effects of mitochondrial involvement in induction of apoptosis are the release of cytochrome c and the mitochondrial membrane
potential (Ceende et al., 1993). My previous studies demonstrated the release of cytochrome c in T. vaginalis-treated RAW264.7 cells. Here, I have shown that significant change of the
membrane potential was observed in T. vaginalis-treated RAW264.7 cells (Figure II-1B). Moreover, in Bcl-2-transfected RAW264.7 cells, overexpression of Bcl-2 not only did not
inhibit T. vaginalis-induced cytochrome c release and Bax activity (Figure II-4A), but also did not alter elevation of the membrane potential in T. vaginalis-treated RAW264.7 cells (Figure
of its localization at mitochondrial membrane (Cuende et al., 1993- Zhu et al., 1996). Therefore, my findings indicate that T. vaginalis-induced apoptosis in RAW264.7 cells may be
independent of Bcl-2 expression. In addition, it is possible that the inability of Bcl-2 overexpression to inhibit T. vaginalis-induced apoptosis may be induced by other Bcl-2 family
rather than Bcl-2 dependent apoptotic pathways.
In addition to mitochondrial involvement, I also examined the relationship between Bcl-2
family activity and the cleavage of caspases, a key event of apoptotic pathway in T. vaginalis-treated RAW 264.7 cells. A family of caspases related to the C. elegans CED-3 plays a central
role in driving the apoptotic pathways triggered by a variety of stimuli. (Ellis et al., 1986). Activation of caspases through transmission of diverse apoptotic signals leads to cleavage of
target proteins and execution of the apoptotic program (Los et al., 1999). Recent study has shown that overexpressed Bcl-2 can dramatically inhibit the change of mitochondria
membrane potential and activation of both caspase-9 and –3, and thus Bcl-2 overexpression inhibits TCHQ-induced apoptosis in NIH3T3 cells (Lin et al., 2004). Not expected, in this
study, the cleavages of pro-caspase-9, -3 and PARP was not inhibited by overproduction of Bcl-2 (Figure II-4C). This finding supports a hypothesis that T. vaginalis-induced apoptosis
showed that although a decrease in the ratio of Bcl-2 to Bax was not apparent, the expression level of Bcl-xL dramatically was decreased with the culture time (Figure II-2, 5A). These
results are consistent with previous observation, which that only Bcl-xL, and not Bcl-2, was
observed to inhibit apoptosis in WEHI-231.7 cells in response to cross-linking of IgM and
other stimuli (Gottschalk et al., 1994). Interestingly, the downregulation of Bcl-xL was almost
abolished by the treatment with both caspase inhibitors (Figure II-5B). Cheng et al. similarly
reported that the anti-apoptotic protein Bcl-2, a homologue of Bcl-xL, serves as a substrate for
caspase-3 in cells undergoing apoptosis and that the cleavage product of Bcl-2 exerts a
death-promoting function (30). In general, caspase-3 activation is though to require Apaf-1 and cytochrome c release from mitochondria. This mechanism is regulated by the Bcl-2 family
(Zou et al., 1997). Thus, my result suggests that caspase-3 may play pivotal role in the process of apoptosis as well as the downregulation of Bcl-xL by T. vaginalis.
Several studies have demonstrated that Bcl-xL can associate with caspase-1, caspase-8, and
caspase-9 in mammalian cells (Chinnaiyan et al., 1997; Hu et al., 1998). Here when Bcl-xL
-overexpressing RAW 264.7 cells are treated with T. vaginalis, I have shown that Bcl-xL
associated with cytochrome c release, caspase-9 and caspase-3 (Figure II-6C, D). Therefore,
even though the involvement of Apaf-1 is unclear, it is possible that Bcl-xL could inhibit T.
vaginalis-induced apoptotic cell death. It might be through alteration of the interaction of
Apaf-1/caspase-pathway (Giri et al., 2003). The protozoan parasite E. histolytica-induced DNA fragmentation is unaffected in caspase 8-deficient Jurkat cells (Huston et al., 2000). In previous my study, I
could not find cleavage of a caspase-8 and Bid activity in T. vaginalis-treated RAW264.7 cells, indicating that the death receptor-mediated apoptotic pathway was not involved in T.
vaginalis-induced apoptosis.
In conclusion, my study suggests that induction of apoptosis by T. vaginalis was found to be
associated with a reduction of expression of anti-apoptotitc Bcl-xL protein, not Bcl-2. In
addition, the activation of caspase-9 suggests that mitochondria play a role in T.
vaginalis-induced RAW264.7 cell death. The downregulation of Bcl-xL expression critically involves
activation of caspase-3, which downregulated Bcl-xL, and thereby provides a positive
feedback mechanisms that may be responsible for the T. vaginalis-induced apoptotic cell death. Therefore, it is plausible that the lack of Bcl-2 protection implies that the functions of Bcl-xL
with caspase-3 may be important to regulate the T. vaginalis-induced apoptotic pathway with biological relevance.
CHAPTER III
Trichomonas vaginalis inhibits proinflammatory
cytokine production in macrophages by suppressing
1. Introduction
Trichomonas vaginalis is a flagellate protozoan parasite, which infects the genito-urinary
tract of humans. It is responsible for trichomoniasis, one of the most common nonviral sexually transmitted disease (STD) in the world (Petrin et al., 1998). The vaginal infection
caused by T. vaginalis is severe due to the various factors involved in the development of infection. It has been associated with adverse outcomes of pregnancy (Cotch et al, 1997),
cervical cancer (Kharsany et al., 1993), and the increase in the transmission of human immunodeficiency virus (HIV) (Draper et al., 1998). It is well known that T. vaginalis adheres
to epithelial cells and survives in reproductive tracts by scavenging nutrients from the host (Alderete et al., 1995). Their detachment from epithelial cell results in the cytological change
observed in trichomoniasis (Gupta et al., 1990). It causes a variety of symptoms from a state of severe inflammation and irritation with a frothy malodorous discharge to a relatively
asymptomatic carrier state (Petrin et al., 1998).
In the female reproductive tract, the mucosal immune system is the first stage of the defense
against pathogenic organisms (Underdown and Schiff, 1986). It is mediated by both innate and adaptive immune responses including the humoral and cell-mediated immunities, leading to
of protozoan infection (Scharton-Kersten et al., 1997). Some clinical trials have been reported by treating econazole in combination with ibuprofen isobuthanolammonium to improve
submucosal macrophage cytotoxicity against T. vaginalis (Martinotti et al., 1983; Drage et al., 2000) indicate that the important role of macrophage as a compartment of host immune
system. However, the role of macrophage in T. vaginalis infection is not clear.
Macrophages play a key role against infection as an innate immune system, and are capable of producing TNF-α and IL-12 (Butcher et al., 2001). TNF-α is a multifunctional cytokine
that trnasduces signals of survival, differentiation, and cellular death in diverse cell types and
that elicits diverse biological events by inducing the expression of various genes (Roulston et
al., 1998). It also has been reported that TNF-α plays a major role in defense against mycobacterial infection (Sugawara et al., 1999). IL-12 production by macrophages is significant in acquiring the potential of directing acquired immunity toward a Th-1 biased
response that is crucial to controlling microbial infections (Hsieh et al., 1993). Nuclear factor-κB (NF-factor-κB) is implicated in the regulation of cell growth, differentiation, inflammatory responses, and apoptosis (Medzhitov et al., 1997; Baeuerle and Henkel, 1994). NF-κB
consists of a family of dimeric factors and is reported to get involved in the transcriptional regulation of many cytokine genes for TNF-α, IL-1β, and IL-12 in response to extracellular
signals (Murphy et al., 1995; Cogswell et al., 1994). In unstimulated cells, NF-κB remains as an inactive form in the cytoplasm in association with the inhibitory family of IκB molecules.
(Michael and Yinon, 2000; Read et al., 1994). Thus, the activation of NF-κB is strongly
related to the production of cytokines and the activation of effector molecules in association
with innate immunity.
In this study, I investigated whether the inflammatory response of macrophages elicited by T.
vaginalis infection can be characterized by NF-κB activation. I show here that T. vaginalis induces rapid NF-κB activation in RAW264.7 macrophage during the early adhesion.
However, the activation was not maintained but resulted in blocking the production of proinflammatory cytokines such as TNF-α and IL-12. Furthermore, T. vaginalis infection
induced a state of non-responsiveness to subsequent stimulation with bacterial LPS. Together, MG-132 blocking IκB-α degradation curtailed apoptotic cell death in cells adhered with T.
vaginalis demonstrates that the NF-κB activation appears to be related to anti-apoptosis. These results suggest T. vaginalis is able to induce inhibitory mechanism to avoid or delay the
2. Materials and Methods
Materials
DMEM (Dulbecco’s modified Eagle’s medium) and FBS (fetal bovine serum) were purchased from sigma (St. Louis, USA). Antibodies for IκB-α and β-actin were purchased
from Santa Cruz Biotechnology (Santa Cruz, USA) and for p50 and p65 from Calbiochem (San Diego, USA). Annexin V/PI kit was purchased from Pharmingen (San Diego, USA).
Parasites
Trichomonas vaginalis strain KT-4 (kindly provided by J. S. Ryu, Department of Parasitology, University of Han-yang, Korea) was used in this study. Trichomonads were
cultured in Diamond's Trypticasc-yeast extract-maltose (TYM) medium (Flynn et al., 1995) and supplemented with 10% heat-inactivated horse serum in 5% CO2 environment for 24 h at
37℃. Cultured parasites were always monitored for motility and only those viable parasites (>99%) were for experiments.
Cell lines culture and in vitro infection
The murine monocyte/macrophage cell line, RAW 264.7, was cultured in DMEM supplemented with 10% FBS, penicillin (100 U/ml), and streptomycin (100 U/ml) in 5% CO2