저작자표시-비영리-변경금지 2.0 대한민국 이용자는 아래의 조건을 따르는 경우에 한하여 자유롭게 l 이 저작물을 복제, 배포, 전송, 전시, 공연 및 방송할 수 있습니다. 다음과 같은 조건을 따라야 합니다: l 귀하는, 이 저작물의 재이용이나 배포의 경우, 이 저작물에 적용된 이용허락조건 을 명확하게 나타내어야 합니다. l 저작권자로부터 별도의 허가를 받으면 이러한 조건들은 적용되지 않습니다. 저작권법에 따른 이용자의 권리는 위의 내용에 의하여 영향을 받지 않습니다. 이것은 이용허락규약(Legal Code)을 이해하기 쉽게 요약한 것입니다. Disclaimer 저작자표시. 귀하는 원저작자를 표시하여야 합니다. 비영리. 귀하는 이 저작물을 영리 목적으로 이용할 수 없습니다. 변경금지. 귀하는 이 저작물을 개작, 변형 또는 가공할 수 없습니다.
Doctoral Thesis in Natural Sciences
Mechanistic Study on the Therapeutic Strategy
to Enhance Glioma Cell Death
through Inhibition of Proteasome and Lysosome
Ajou University Graduate School
Department of Biomedical Sciences
Mechanistic Study on the Therapeutic Strategy
to Enhance Glioma Cell Death
through Inhibition of Proteasome and Lysosome
Kyeong Sook Choi, Advisor
I submit this thesis as
the Doctoral thesis in natural sciences
June, 21th, 2016
Ajou University Graduate School
Department of Biomedical Sciences
The Doctoral thesis of In Young Kim in natural sciences is hereby approved.
Thesis Defense Committee President
Yup Kang Seal
Member
Kyeong Sook Choi Seal
Member
Jong-Soo Lee Seal
Member
You-Sun Kim Seal
Member
Taeg Kyu Kwon Seal
Ajou University Graduate School
i ABSTRACT
-Mechanistic Study on the therapeutic Strategy to Enhances Glioma Cell Death
through Inhibition of Proteasome and Lysosome
Malignant gliomas, the most common brain tumors with high mortality, remain largely incurable despite multimodal treatments including surgical resection, radiotherapy, and chemotherapy. Thus, the researchers are currently attempting to develop novel therapeutic strategies for malignant gliomas. Recent studies show that dysregulation of protein maintenance has been considerably linked to many diseases. Therefore, protein degradation system has become a platform for drug targeting, and mechanism-based drugs are currently developed. Bortezomib (BZ), a proteasome inhibitor, and chloroquine (CQ), a lysosomotropic agent, can disrupt cellular protein homeostasis (proteostasis) via interruption of protein degradation systems, including proteasome and lysosome. In part I, we investigate the role of eIF2a in BZ-induced cell death. We found that ER-derived dilation preceded bortezomib induced cell death. eIF2α was initially phosphorylated but it was dephosphorylated in parallel with the ER dilation. Overexpression of eIF2a WT markedly increased rate of phosphorylated eIF2a (exogenous) and blocked bortezomib-induced ER vacoulation and cell death. In contrast, overexpression of dominant negative eIF2a S51A does not affect rate of phosphorylated eIF2a and cell death. These results suggests that dephosphorylation of eIF2a may be important for the ER dilation and cytotoxic effect of bortezomib. These results suggest that eIF2a dephosphorylation rather than its phosphorylation may be important for the cytotoxic effect of bortezomib. In addition, knockdown of eIF2a sensitized bortezomib-induced cell death via increasing caspase-dependent apoptosis. Collectively, regulation of eIF2a phosphorylation and expression may provide a therapeutic strategy to sensitize glioma cell to bortezomib-mediated cell death. We found that combined treatment
ii
-with bortezomib and salubrinal, an inhibitor of GADD34-PPiC phosphatase complex, very effectively induced the dilation of the endoplasmic reticulum and subsequent cell death. Interestingly, either salubrinal or bortezomib increased the phosphorylation levels of eIF2α, but combined treatment markedly reduced them. In addition, co-treatment with salubrinal decreased expression levels of eIF2a. Taken together, these results suggest that salubrinal effectively overcomes the resistance of malignant glioma cells to BZ, via dependently- and independent-manner of eIF2a phosphorylation.
In part II, we investigate whether co-treated phytochemicals sensitized CQ-induced cell death in glioma cells. We show that treatment with subtoxic doses of CQ, when combined with kaempferol, a flavonoid, effectively induces cell death in various glioma cells, but not in normal astrocytes, suggesting that the combined regimen using kaempferol and CQ may provide a safe therapeutic strategy to selectively kill resistant glioma cells. The cell death induced by kaempferol and CQ in U251MG cells was partially dependent on caspase-mediated apoptosis and accompanied by mitochondrial dysfunction, ER stress, and DNA damage. We found that kaempferol treatment increased the numbers of lysosome, whereas CQ treatment increased the lysosomal masses leading to their swelling. Combined treatment with kaempferol and CQ further increased the lysosomal masses, but culminated in the disruption of lysosomal compartments. While kaemfperol treatment increased the active mature forms of cathepsin D, CQ treatment markedly blocked the processing and activity of cathepsin D. Interestingly, combined treatment induced the release of unprocessed cathepsin D into the cytosol and the recovery of its activity. Knockdown of cathepsin D significantly attenuated the cell death induced by kaempferol and CQ, suggesting the functional involvement of these released cathepsin D proteins in this cell death. Taken together, our results suggest that the membrane destabilization of accumulated lysosomes and the resultant release of lysosomal proteases may critically contribute to the irreparable damage of various
iii
-organelles and glioma cell death by kaempferol plus CQ.
In this study, we show that salubrinal effectively overcomes the resistance of malignant glioma cells to BZ, via dependently- and independent-manner of eIF2a phosphorylation. Also, we show that combined treatment with kaempferol and CQ effectively induces cell death in glioma cells, via lysosomal overloading and rupture. Collectively, these results suggest that proteostasis perturbation by targeting of UPR components or lysosome may provide potential therapeutic effect for targeting malignant glioma cells over normal cells.
Keyword: proteostasis, protein degradation, bortezomib (BZ), chloroquine (CQ),
iv
-TABLE OF CONTENTS
ABSTRACT ···i
TABLE OF CONTENTS ···iv
LIST OF FIGURES ···xi
I. INTRODUCTION ···1
II. MATERIALS AND METHODS ···24
A. MATERILAS ···24
1. Chemicals ···24
2. Antibodies ···24
B. METHODS ···25
a. Culture of glioma cell lines and normal human astrocytes ···25
b. Determination of cellular viability using calcein-AM and EthD-1 (Live-Dead assay) ···25
c. Determination of cellular viability by an MTT assay ···26
d. Determination of synergism by the combination inducx (CI) method ···26
e. Colonogenic cell survival assay ···26
f. Western blotting ···27
g. Establishment of the stable cell lines expressing the fluorescence specifically in endoplasmic reticulum ···27
h. Immunocytochemisty ···28
i. small interfering RNAs ···28
j. Knockdown experiments using small hairpin (sh) RNA ···29
k. Assay of aggresome formation ···29
l. Visualization of autophagic vacuoles ···29
m. LysoTracker Red staining ···30
n. Detection of acidic vesicular organelles (AVOs) with acridine orangs staining ···30
o. BODIPY FL-pepstatin staining ···30
p. HCS LipidTOXTM neutral lipid staining ···30
v
-r. Intracellular pH detection ···31
s. Transmission electron microscopy ···31
t. Measurement of mitochondrial superoxide production ···32
u. Statistical analysis ···32
PART1.eIF2a modulates bortezomib-induced vacuolation and cell death in glioma cells via phosphorylation-dependent and independent manner ···33
1. INTRODUTION ···34
2. RESULTS ···40
2.1. Proteasome inhibitors indue non-apoptotic cell death in glioma cell death in glioma cells accompanying ER-derived vacuolation ···40
2.2. The eIF2a phosphorylation at serin 51 blocks bortezomib-induced vacuolation and cell death in glioma cells ···47
2.3. Protein synthesis may play an important role in ER vacuolation and cell death by bortezomib ···52
2.4. Dephosphorylation of eIF2a rapidly accumulates aggregates in cells, leading to cell death by bortezomib ···56
2.5. The eIF2a downregulation sensitizes glioma cells to bortezomib-induced apoptosis ···58
2.6. Dephosphorylation and downregulation of eIF2a may be responsible for the synergistic effect of salubrinal and bortezomib on ER-derived vacuolation and cell death ···61
2.7. Protein synthesis may play an important role in ER vacuolation and cell death by salubrinal and bortezomib ···69
2.8. Bortezomib-induced poly-ubiquitination of proteins in further enhanced but ubiquitinated protein aggregates are dispersed by co-treatment with salubrinal ···72
2.9. Human astrocytes are resistant to the combined treatment with salubrinal and bortezomib ···75
3. DISCUSSION ···77
PART2. Flunarizine overcomes the resistance of glioma cells to bortezomib via induction of paraptosis-like cell death ···84
vi
-2. RESULTS ···87 2.1. Flunarizine sensitizes glioma cells, but not normal astrocytes,
to bortezomib-mediated non-apoptotic cell death ···87 2.2. The combined treatment with flunarizine and bortezomib induces the swelling
of ER in glioma cells ···93 2.3. CHOP and NOXA is critically involved in the cell death by flunarizine plus
bortezomib ···99 2.4. Flunarizine plus bortezomib induces paraptosis-like cell death ···101 2.5. Critical role of ROS in the cell death by the combined treatment with
flunarizine and bortezomib ···103 2.6. Critical role of intracellular Ca2+ in the cell death by the combined treatment with flunarizine and bortezomib ···105 2.7. Disruption of aggresome formation may be involved in the cell death induced by flunarizine and bortezomib ···107 3. DISCUSSION ···110 PART3. Combination of chloroquine and kaempferol induces cell death
via lysosomal overloading and rupture ···115 1. INTRODUTION ···116 2. RESULTS ···119 2.1. Kaempferol sensitizes human glioma cells to chloroquine-mediated cell death,
but not normal astrocytes ···119 2.2. Kaempferol promotes autophagy in glioma cells ···123 2.3. The sensitizing effect of chloroquine on kaempferol-mediated cell death may
be associated with its activity as the lysosomal inhibitor ···128 2.4. Co-treatment with kaempferol disrupts chloroquine-induced swollen lysosomal
structures ···133 2.5. Cathepsin D released from the disrupted ysosomes may contribute to the cell death by the combined treatment with kaempferol and chloroquine ···139 2.6. Lysosomal rupture by combined treatment with kaempferol and chloroquine
injures cellular organelle including mitochondrial and DNA ···146 3. DISCUSSION ···151 IV. CONCLUSION ···159
vii
viii
-LIST OF FIGURES
Fig. 1. Protein fates in the proteostasis network ···8
Fig. 2. The unfolded protein response ···9
Fig. 3. Two systems for protein degradation within cells ···11
Fig. 4. Proteasome inhibitors induce preferential cell death in malignant glioma cells, but not human normal astrocytes ···42
Fig. 5. Proteasome inhibitors induce non-apoptotic cell death in glioma cells ···43
Fig. 6. Proteasome inhibitors induce cytoplasmic vacuolation in glioma cells ···44
Fig. 7. Proteasome inhibitors induce ER-derived vacuolation in glioma cells ···45
Fig. 8. Proteasome inhibitors induce ER stress in glioma cells ···46
Fig. 9. Overexpression of eIF2a WT blocked cell death by bortezomib ···49
Fig. 10. While overexpression of eIF2a WT blocked borteozmib-induced cell death, overexpression of eIF2a S51A accelerated bortezomib-induced cell death ··· 50
Fig. 11. In cells overexpressing eIF2a S51A, bortezomib induced non-apoptotic cell death ···51
Fig. 12. Decrease of misfolded proteins inhibits the cell death by overexpression of eIF2a WT ···54
Fig. 13. eIF2a dephosphorylation accelerates bortezomib-induced aggresome formation ···57
Fig. 14. knockdown of eIF2a sensitized cell death, but it did not affect ER vacuolation by bortezomib ···59
Fig. 15. Knockdown of eIF2a increased apoptosis by bortezomib ···60
Fig. 16. Salubrinal plus bortezomib synergistically induces cell death in human glioma cells ···64
Fig. 17. Salubrinal accelerates bortezomib/MG132-induced ER dilation ···66
Fig. 18. Combined treatment with salubrinal and bortezomib partly induces apoptosis ···67
Fig. 19. Status of eIF2a in T98G cells treated with salubrinal and/or bortezomib ···68
Fig. 20. Inhibition of translation decreases the cell death and ER vacuolation by salubrinal plus bortezomib in T98G cells ···70
ix
-Fig. 21. Chemical chaperons partially inhibit the cell death by salubrinal plus borteozmib
in T98G cells ···71
Fig. 22. Salubrinal disrupts aggresome formed by bortezomib ···74
Fig. 23. Salubrinal plus bortezomib did not induce cell death and alter cell morphology in human normal astrocytes ···76
Fig. 24. Flunarizine plus borteozmib synergistically induces cell death in human glioma cells ···89
Fig. 25. Flunarizine plus bortezomib did not alter cell morphology and induce cell death in human normal astrocytes ···90
Fig. 26. Combined treatment with flunarizine and bortezomib induces non-apoptotic cell death ···91
Fig. 27. Flunarizine accelerates bortezomib-induced ER dilation ···97
Fig. 28. Combined treatment with flunarizine and bortezomib induces ER stress in glioma cells ···98
Fig. 29. CHOP and NOXA is critically involved in cell death induced by flunarizine plus bortezomib ···100
Fig. 30. Combined treatment with flunarizine and bortezomib induces paraptosis-like cell death in T98G cells ···102
Fig. 31. Combined treatment with flunarizine and bortezomib dramatically increases the ROS levels in glioma cells ···104
Fig 32. Combined treatment with flunarizine and bortezomib increases the intracellular Ca2+ levels in glioma cells ···106
Fig. 33. Flunarizine disrupts aggresome formed by bortezomib ···108
Fig. 34. CHOP and NOXA induction did not affect the accumulation of ubiquitin -conjugated proteins in glioma cells ···109
Fig. 35. Combination of kaempferol and CQ effectively induces glioma cell death, sparing normal astrocytes ···121
Fig. 36. z-VAD-fmk, but not necrostatin-1, partly inhibits the cell death by kaemfperol and chloroquine ···122
Fig. 37. Kaempferol induces autophagy ···125
Fig. 38. Kaemfperol promotes autopahgic flux ···126
x
-Fig. 40. Combined treatment with kaemfperol and PI3K inhibitors does not induce cell death in U251MG cells ···129 Fig. 41. Suppression of autophagy does not induce the cell death in U251MG cells,
when combined with kaempferol ···130 Fig. 42. Combined treatment with kaempferol and drugs inhibiting lysosomal degradation
effectively cell death in U251MG cells ···131 Fig. 43. Combined treatment with kaempferol and other anti-malaria drug significantly
cell death in U251MG cells ···132 Fig. 44. Combined treatment with kaempferol and chloroquine disrupts lysosomal
structures ···136 Fig. 45. Kaemfperol plus chloroquine accumulates protein aggregates in lysosome ··· 137 Fig. 46. Kaempferol plus chloroquine accumulates neutral lipid in lysosome ···138 Fig. 47. Combined treatment induces the release of unprocessed cathepsin D into the
cytosol ···142 Fig. 48. Cathepsin D released from lysosome by kaempferol plus chloroquine has
the activity in cytosol ···143 Fig. 49. Knockdown of Cathepsin D blocks the cell death by combined treatment
with kaempferol and chloroquine ···144 Fig. 50. Combined treatment with kaempferol and chloroquine decreases cytosolic pH
···145 Fig. 51. Kaempferol plus chloroquine injures mitochondria in U251MG cells ···148 Fig. 52. Kaempferol plus chloroquine increases DNA damage ···149 Fig. 53. A schematic diagram of cell death induced by the combined treatment with
1
I. INTRODUCTION
A. Malignant glioma
Malignant gliomas are the most common adult primary brain tumor. They are characterized by local proliferation, insidious infiltration throughout the brain parenchyma, and robust angiogenesis (Chen et al., 2011a). Malignant gliomas are classified by the World Health Organization (WHO) as either grade III/IV tumors such as anaplastic astrocytoma, anaplastic oligodendroglioma, anaplastic oligoastrocytoma and anaplastic ependymomas, or grade IV/V tumors, such as glioblastoma multiforme (GBM). GMBs usually follow an aggressive course, and patients have a median survival of 8 months to 10 months (Curran et al., 1993). Currently, the only known risk factor for the development of malignant glioma is exposure to ionizing radiation, and no other environmental exposures including cell phone use, infection of trauma has been shown to have an effect (Butowski, 2015). The three main tenants of treatment for high grade gliomas have remained consistent for the past 3 decades: microscope aided surgical resection, internal and external radiation therapy, and local and systemic chemotherapy. Several chemotherapeutic agents, such as temozolomide (TMZ), cisplatin, carmustine, and lomustine, have been used to slow the progression of these incurable cancers. However, the resistance of gliomas to current treatment protocols still remains a major concern in cancer therapy.
2
B. Advantages of combination treatment in cancer therapy
According to the GLOBOCAN project by World Health Organization, globally in 2012, 14.1 million new cancer cases were diagnosed, 8.2 million people died from cancer and 32.6 million people were five-year cancer survivors. By 2025, 19.3 million new cancer cases are expected to be diagnosed each year (http://globoca.iarc.fr/Pages/fact_sheets_cancer.aspx, IARC. Estimated Cancer Incidence, Mortality and Prevalence Woldwide in 2012). The risk of cancer increases significantly with age and many cancers occur more commonly in developed countries (Bernard W. Stewart and Christopher P. Wild, World Cancer Report 2014). Because of the aging of the world population and increasing adoption of cancer-causing behaviors, the global burden of cancer continues to rise largely (Jemal et al., 2011).
In current cancer therapy, there are many treatment options including surgery, chemotherapy, radiation therapy, hormonal therapy, and targeted therapy. The selection of therapy depends upon the location and grade of the tumor, as well as the general state of the patient. The ideal goal of treatment is complete removal of the cancer without damage on the body. Sometimes this can be accomplished by surgery, but the propensity of cancers to invade adjacent tissue or to spread to distant sites by microscopic metastasis often limits its effectiveness; and chemotherapy and radiotherapy can have a negative effect on normal cells (Enger, Eldon; et al.Concepts in Biology' 2007 Ed.2007 Edition. McGraw-Hill. p. 173.ISBN978-0-07-126042-8. Retrieved23 November2012. ).
Chemotherapy is the one of major methods for cancer treatment, however, its failure is frequent due to the cytoxicity of the drug and the acquired resistance to the drug (De Souza et al., 2011; Liu et al., 2015a). In addition, most drugs for chemotherapy are non-specific and they have a wide-range of side-effect (Persidis, 1999; Crawford, 2013). To resolve the problems involved in use of chemotherapeutic drugs, researcher have modified treatment protocols that
3
a combination of drugs with different molecular targets is used (Rahman and Hasan, 2015). This combination strategy has an advantage in reducing the risk of clonal selection of resistant tumor cells to one drug only. Moreover, each drug can be applied at a lower dose to minimize side-effects in combination therapy (Cimino et al., 2013; Rahman and Hasan, 2015). Actually, in order to prevent drug resistance in cancer, combination therapy is believed to be more effective than sequential treatment (Bozic et al., 2013; Komarova and Boland, 2013). Therefore, co-administration of cytotoxic agents (such as paclitaxel, irinotecan and 5-FU) and agents inferring growth signals (including VEGFR, EGFR, MPK, PI3K or mTOR pathway) has been tested clinically for improved antitumor efficacy (Li et al., 2014). In recent years, drugs targeting the biological process, such as bortezomib (proteasome inhibitor), tanespimycin (heat shock protein 90 inhibitor) and AMG655 (death receptor 5 antibody), have attracted more and more attention as a candidate for combined treatment (Li et al., 2014).
4 C. Proteostasis
Cells must constantly maintain the homeostasis of proteins in various environment. Proteostasis, a mixed word using protein and homeostasis, is the process that controls the synthesis, folding, trafficking and degradation of proteins within cells and it is directly involved in the functional maintenance of protein (Dufey et al., 2015). Proteostasis can be changed by perturbations of physiological/pathological factors and one of altering proteteostasis it the failure of protein folding. Various stress conditions such as oxidative stress, inappropriate temperature and metabolites can increase unfolded and misfolded proteins. Particularly, oxidative stress can alter the protein via a mutation of gene or an aberrant posttranslational modification such as disulfide formation (Powers et al., 2009). In addition, breakdown of the refolding machinery including chaperone proteins and folding factors including disulfide bridges accumulates misfolded proteins.
For survival, cells must maintain the balance between protein production and degradation (Hede et al., 2014). The degradation process is important not only for normal cell growth and development but also for the adaptive responses to stresses such as starvation and infection. In addition, malfunctioning of degradation system increases the old, misfolded, or damaged proteins and form large aggregates (Kraft et al., 2010), leading to disruption of proteostasis. In particular, suppression of the degradation systems including proteasomes is the one of causes to perturb protein homeostasis. The Ubiquitin-proteasome system (UPS), is responsible for degradation 80-90% of proteins including many regulated, short-lived, abnormal, denatured, and damaged proteins (Rock et al., 1994). By contrast, autophagy, which is primarily responsible for the degradation of most long-lived proteins, degrades not only aggregated proteins but also cellular organelles (mitochondria, peroxysomes, ribosomes, infectious organisms) (Lilienbaum, 2013).
5
Dysregulation of protein maintenance has been considerably linked to many diseases including cystic fibrosis (loss-of-function diseases), Alzheimer’s, Parkinson’s, and Huntington’s disease (gain-of toxic-function diseases), among other diseases (Powers et al., 2009). In addition, protein homeostasis is also important for the cancer development process. Because cancer cells require the increased protein synthesis for continuous division, proteostasis networks including chaperones and degradation systems (Fig. 1) must be strictly regulated in cancer cell. Moreover, since genomic alterations are a characteristic of the cancer cells, the point mutations in protein coding sequences often result in the accumulation of misfolded protein (Vogelstein et al., 2013). This results in an imbalance when the degradation load exceeds the capacity of the UPS. Thus, genomic alterations may make cancer cells more dependent than normal cells on mechanisms of proteostasis, including protein folding and degradation (Deshaies, 2014). Actually, these features make cancer cell sensitive to drugs that inhibit chaperones and disrupt proteostasis, such as Hsp90 inhibitors or proteasome inhibitors. Since the endoplasmic reticulum (ER) is responsible for protein synthesis, folding and secretion, it is the most important organelle to maintain proteostasis. At the ER, a complex network including chaperones molecules, unfolded protein response (UPR) and ER-associated degradation (ERAD) mechanism makes the efficient production of functional proteins (Araki and Nagata, 2011). An accumulation of misfolded protein in the ER, a condition called “ER stress”, indicates the change of proteostasis and it triggers the UPR, an adaptive mechanism. In tumor cells, excessive proliferation induces the alteration of microenvironment, including hypoxia, nutrient deprivation and acidosis, and results in ER stress (Ma and Hendershot, 2004). This stress condition activates the UPR, acting as an accommodating factor to stress for promoting cell survival. Thus, UPR in cancer cells increases aggressiveness and resistance to chemotherapeutic agents in cancer cells (Clarke et al., 2014; Wang and Kaufman, 2014). For
6
this reason, the drugs targeting UPR components are recently attracted as potential anti-cancer therapeutics.
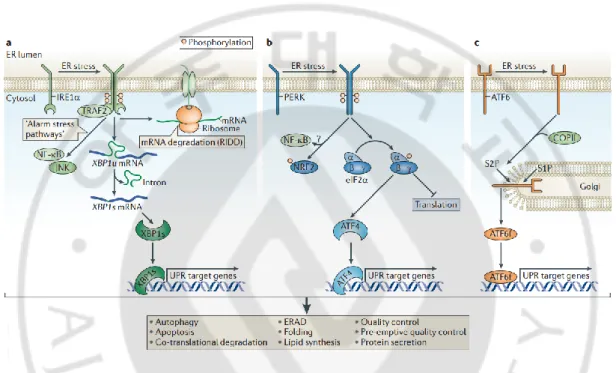
The UPR have three types of stress sensors, including PKR-like ER kinase (PERK), Inositol-requiring enzyme 1 (IRE1) and Activating transcription factor 6 (ATF6) (Fig. 2). They are located at the ER membrane and convey information about the protein-folding status to the nucleus and cytosol for decision of cell fate under ER stress (Hetz, 2012). When misfolded proteins are increased in the ER, the molecular chaperone Bip/Grp78 is dissociated with PERK, IRE1 and ATF6 and binds to misfolded proteins. Separation of Bip/Grp78 triggers activation of three stress sensors through phosphorylation, homo-dimerization and translocation. Activated PERK phosphorylates eukaryotic translation inhibition factor α (eIF2α) serine 51 leading to inhibition of global protein translation. Also, phosphorylation of eIF2α increases the selective mRNA translation of Activating transcription factor 4 (ATF4), playing a role as a controller for expression of UPR-target genes involved in amino acid metabolism, antioxidant responses, autophagy and protein folding (Harding et al., 2003). Increased expression of ATF4 regulates CHOP/GADD153, a transcription factor that participate in the core mitochondrial apoptosis pathway through the control of the expression of BCL-2 family members (Urra et al., 2013). IRE1 triggers UPR via a cytoplasmic kinase domain and an RNAse domain (Hetz and Glimcher, 2009; Hetz et al., 2011; Hetz, 2012; Chen and Brandizzi, 2013). ER stress conditions induce dimerization and auto-transphosphorylation of IRE1, resulting in a conformational change in the cytosolic domain and activation of RNase domain (Walter and Ron, 2011). Active IRE1 induces the expression of XBP1s through the unconventional splicing of the transcription factor X-box binding protein (XBP1) mRNA (Yoshida et al., 2001; Calfon et al., 2002; Lee et al., 2002). Increased XBP1s modulates the expression of UPR target genes including ER chaperones, glycosylation enzymes, ER-associated degradation (ERAD) and
7
components of the ER translocon (Lee et al., 2003; Acosta-Alvear et al., 2007). In addition, IRE1 targets other mRNAs and microRNAs via a process named Regulated Ire1-dependent decay (RIDD) (Maurel et al., 2014). Finally, during the ER stress, ATF6 is transported to the nucleus from ER by way of Golgi apparatus (Haze et al., 1999; Adachi et al., 2008). Translocated ATF6 regulates the expression of genes associated with ERAD and enhances the expression of XBP1 mRNA (Adachi et al., 2008). Each sensor plays an important role involved in cell adaptation response for protein folding and determination of cell fate.
8
9
Fig. 2. The Unfolded Protein Response (UPR) (Hetz, 2012). Three types of stress sensors,
(A) Inositol-requiring enzyme 1 (IRE1), (B) PKR-like ER kinase (PERK),) and (C) Activating transcription factor 6 (ATF6).
10 D. Protein degradation system
Protein degradation is essential for the life and death of every cell. Cells remove the old, misfolded, mutated, or otherwise damaged proteins by protein degradation system. Degradation of intracellular proteins is associated with management of cellular processes, such as cell cycle and division, regulation of transcription factors, and assurance of the cellular quality control. Thus, breakdown of proteins occurs constantly inside cells as the production of new proteins must be balanced by an equal amount of protein degradation for cells to survive (Hede et al., 2014). Interestingly, aberration of protein degradation system has been implicated in the pathogenesis of many human diseases such as cancer and neurodegenerative disorders (Ciechanover, 2013). As a result, protein degradation system has become a platform for drug targeting, and mechanism-based drugs are currently developed. In eukaryotic, two major pathways for protein degradation are the ubiquitin-proteasome system (UPS) and lysosomal proteolysis (Fig. 3).
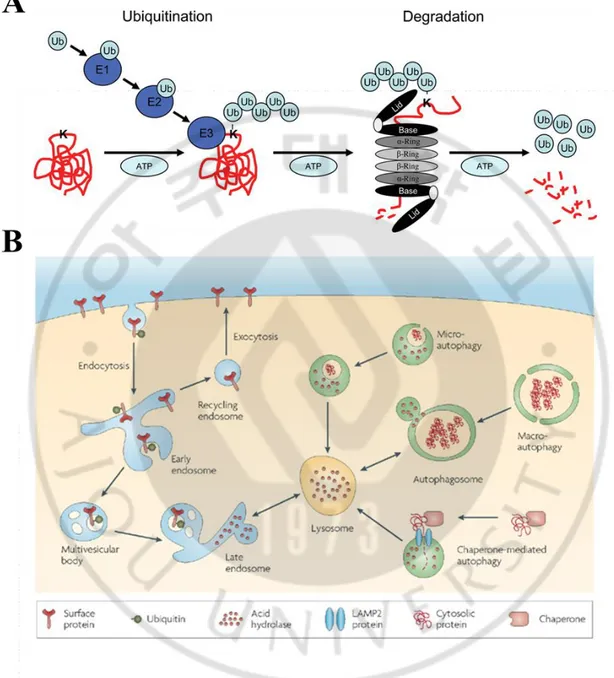
11
Fig. 3. Two systems for protein degradation within cells (Tai and Schuman, 2008). (A)
12 a. Ubiquitin-Proteasome system (UPS)
The UPS is a highly complex network. It maintains protein homeostasis and cell viability through the selective turnover of targeted substrates such as short-lived, abnormal, denatured, or damaged protein (Ciechanover, 2005). Protein degradation by the UPS is composed two major steps: 1) polyubiquitination of the target protein and 2) proteolytic degradation by proteasome complex (Orlowski and Wilk, 2000; Ciechanover, 2005; Shen et al., 2013).
Polyubiquitination is defined as the binding of many ubiquitin molecules to the target protein. Ubiquitin is a small regulatory protein consisted of 76-amino acid unique, and the conjugation of ubiquitin to substrates is associated with a variety of biological processes ranging from proteolysis to DNA damage tolerance (Li and Ye, 2008). The mechanism of ubiquitin conjugation reactions requires a set of enzymes that includes an activating enzyme (E1), a conjugating enzyme (E2), and a ligase (E3). E1 enzymes activate ubiquitins using ATP. As a result, the carboxyl group of an ubiquitin is covalently ligated to the active cysteine residue of the E1 enzyme. Subsequently, the ubiquitin is transferred to the active cysteine residue in an E2 enzyme that in turn relays the ubiquitin molecule to a substrate in the presence of an E3 ligase (Pickart, 2001; Weissman, 2001). These steps facilitate binding of ubiquitin to lysine residues on the target protein. After monoubiquitination of the substrate, the process is repeated to form an elongated chain of ubiquitin residues. Proper recognition of ubiquitinted substrates by the proteasome complex is thought to require a minimum of four ubiquitin residues in the polyubiquitin chain (Johnson, 2015). After linkage to one or more sites in the substrate (mono- or multi-ubiquitylation) ubiquitin can use one of its 7 internal lysine residues (K6, K11, K27, K29, K33, K48 and K63) or its N-terminus to form ubiquitin chains (poly-ubiquitylation) (Kulathu and Komander, 2012). Ubiquitin chains have distinct structures and properties; consequently they have a different impact on the fate of the target protein. For example
K48-13
linked chains result in proteasomal degradation, whereas K63-linked chains are involved in cellular signaling (Kulathu and Komander, 2012). In addition, ubiquitylated proteins can be recognized by chain specific ubiquitin-binding proteins (“readers”) (Husnjak and Dikic, 2012).
Proteins tagged with ubiquitin are recognized and degraded by the 26S proteasome complex (Gallastegui and Groll, 2010). The 26S proteasome complex is a non-lysosomal proteolytic machine that is localized in the cytosol and nucleus of eukaryotic cells (Bhaumik and Malik, 2008). It consists of a 20S catalytic core that is capped at both ends by two 19S regulatory subunits forming lid. The 19S subunits act as recognition and entry site for protein (Peters et al., 1993). Following recruitment to the proteasome, ubiquitin polypeptides are removed from target proteins by a family of deubiquitinating enzymes (DUBs), some of which are associated with the 19S lid. The 19S base component plays a key role in unfolding of the substrate protein and delivery of the unfolded, deubiquitinated substrate into the 20S catalytic core particle (Johnson, 2015). The 20S core resembles a cylinder composed of two α and two β rings (Groll et al., 1997; Bhaumik and Malik, 2008). The α rings are non-catalytic and guard the entrance of unfolded proteins to the active site of the complex by allowing access to only unfolded proteins (Groll et al., 1999). The β rings serve proteolytic activities (chymotrypsin-like, trypsin-like and post-acidic of caspase-like) through each component including β5, β2 and β1 (Frankland-Searby and Bhaumik, 2012). Collectively, these subunits degrade substrate proteins into short oligopeptides.
Because the ubiquitin-proteasome system is a common mechanism for degradation of intracellular proteins, it plays important roles in various cellular process including cell cycle progression, DNA repair, signaling pathways, transcription, cell stress response, and apoptosis. Thus, deregulation of ubiquitin-proteasome system is associated with various diseases, e.g. cancer. It has been demonstrated that malignant cells harbor elevated proteasome activity
14
compared with normal cells (Arlt et al., 2009; Ma et al., 2009). Besides solid tumor cells, it has been reported that expression of proteasome and the mRNA levels are consistently increased to much higher levels in a variety of malignant human hematopoietic cell lines compared with peripheral lymphocytes and monocytes from healthy adults (Kumatori et al., 1990). Cancer cells are more dependent on proteasome activity for their survival and drug resistance; therefore, malignant cells should be more sensitive to treatment with proteasome inhibitors than normal cells. This hypothesis has been supported by many studies (Soligo et al., 2001; Chen et al., 2005; Ovaa et al., 2005; Bazzaro et al., 2006; Chen et al., 2006). Additionally, in cancer cells, the stability and the balance between oncoproteins and tumor suppressor proteins are disturbed in part due to deregulated proteasome-mediated degradation (Liu et al., 2015b). Furthermore, compounds that modify of the ubiquitin-proteasome function provide therapeutic strategies to prevent cancer development and to cure cancer.
15 b. Lysosomal proteolysis (autophagy)
Lysosome is an organelle enclosed by a single membrane and contains various hydrolytic enzymes. It maintains low internal pH (~pH 5) that is the best condition for operating of digestive enzymes through ATP-driven H+ pump in the membrane (Molecular biology of the
cells, 6th Edition). Lysosome puts a termination to several degradation process such as
endocytosis, phagocytosis and autophagy (macro-, micro and chaperone-mediated autophagy (CMA)) (Fig. 3). Lysosome degrades a plethora of compounds, including surface receptors, macromolecules, organelles, pathogens and lipids (Boya, 2012). The degradation process is important during normal cell growth and in development, where it helps restructure differentiating cells, but also in adaptive responses to stresses such as starvation and infection. Thus, lysosomal dysfunction has acute impact on cell homeostasis, resulting in manifold pathological situations, including cancer, infectious diseases, neurodegeneration, and aging. In addition, lysosomes participate in various events associated with neoplastic phenotype and the acquisition of invasive and metastatic potentials (Ubah and Wallace, 2014). In cancer cells, the lysosome are more numerous, larger and have greater cathepsin activity than those in normal cells and the release of cathepsins from cancer cell lysosomes into the extracellular space can promote tumor progression (Mohamed and Sloane, 2006). In addition, the release of certain cathepsins from the lysosome into the cytoplasm is involved in the cell death (Boya and Kroemer, 2008). This release occurs by a process known as lysosome membrane permeabilization (LMP), which possibly occurs following certain changes to the composition of membrane lipids and major lysosomal membrane proteins (Fehrenbacher et al., 2008). LMP can be induced by various stimuli, including reactive oxygen species and endogenous apoptotic stimuli.
16
to lysosome. Autophagy is characterized by the formation of double-membrane structures known as autophagosomes, which sequester cytosolic proteins and organelles. The cargo-loaded autophagosomes then fuse with lysosomes to form autolysosomes in which the engulfed contents are digested by acidic lysosomal hydrolases. Alteration in some of these pathways may have important implication in diseases such as cancer and neurodegeneration. Recently Gewirtz (2014) proposed that that in response to chemotherapy or radiation, autophagy has at least 4 functional forms, including cytoprotective, cytotoxic, cytostatic and nonprotective, (Gewirtz, 2014) and each form of autophagy has different characteristics. For several decades, many researchers have focused on the therapeutic strategy to inhibit autophagy in the belief that autophagy is usually cytoprotective. However, recent many reports show that the cellular responses to chemotherapy or radiotherapy are different depending on tumor types, stages and drugs. Therefore, autophagy-inhibiting strategy may be just one way to enhance the anti-cancer effects, because the autophagy is not of necessity cytoprotective in function. Thus, to sensitize malignancies to chemotherapy and radiotherapy by modulation of autophagy, other functions of autophagy should also be considered.
17 E. Bortezomib
Bortezomib (velcade or PS-341), inhibits the chymotrypsin-like activity of the proteasome through binding with active site in the β5-subunit (Crawford et al., 2006; Chen and Dou, 2010). It is the first proteasome inhibitor approved in the U.S. Food and Drug Administration (FDA) for treatment of multiple myeloma (MM) and mantle cell lymphoma (MCL) (Orlowski et al., 1998; Kane et al., 2003). Bortezomib is clinically used as a single agent and in combination with other conventional anticancer drugs such as dexamethasone in multiple myeloma (Jagannath et al., 2005). Although the mechanisms of anticancer activity by bortezomib are not completely understood, they may include induction of apoptosis, disruption of cell cycle progression, inhibition of proliferation, and anti-angiogenesis (Boccadoro et al., 2005). Bortezomib has also been tested in clinical trials against solid tumors (Papandreou et al., 2004). While the majority of success achieved with bortezomib has been in hematological malignancies, its effect toward solid tumors has been less than encouraging (Chen et al., 2011a). When bortezomib was used as a single agent, a lack of therapeutic effects was observed (Huang et al., 2014). In addition, even when botezomib treatment was combined with several conventional anti-cancer drugs, bortezomib offered no statistically significant response versus these agents alone, suggesting that novel strategies to sensitize solid tumors to bortezomib should be developed. Furthermores, the underlying mechanisms by which solid tumors are resistant to bortezomib should be further investigated.
18 a. Mechanism of action
Until now, various mechanisms of bortezomib to contribute to its anti-cancer effects have been reported from many studies. Bortezomib is known to suppress the NF-κB signaling pathway by preventing proteasomal degradation of inhibitory protein IκB, resulting in the down-regulation of its anti-apoptotic target genes (i.e., IAPs and Bcl-XL) (Adams, 2004). In addition, bortezomib upregulates pro-apoptotic members of the Bcl-2 protein family, including NOXA, BAX and BIK (Oda et al., 2000; Qin et al., 2005; Zhu et al., 2005a; Zhu et al., 2005b; Fribley and Wang, 2006; Perez-Galan et al., 2007; Voortman et al., 2007; Li et al., 2008). Because NOXA is involved in p53-mediated apoptosis and interacts with the anti-apoptotic Bcl-XL, Bcl-2 and NOXA increased by bortezomib induces apoptotic cell death in malignant cells (Adams and Cory, 1998; Gross et al., 1999; Oda et al., 2000). Bortezomib also accumulates unfolded proteins in the ER and cytoplasm, and this condition (ER stress) triggers the generation of reactive oxygen species (ROS), which can result in significant damage to cell structures (Fribley et al., 2004; Obeng et al., 2006; Weniger et al., 2011). Additionally, bortezomib treatment has been shown to upregulate the expression of death receptors DR4 and DR5, leading to enhancement of sensitivity to TRAIL (Nikrad et al., 2005; Liu et al., 2007; Voortman et al., 2007; Shanker et al., 2008; Seki et al., 2010; Yoshiba et al., 2011). Bortezomib has also been shown to activate JNK and to stabilize the cyclin-dependent kinase inhibitors, including p21 and p27, and the tumor suppressor p53 in various malignant cells, resulting in prevention of cell proliferation (Breitschopf et al., 2000; Hideshima et al., 2001; Shah et al., 2001; Hideshima et al., 2003; Williams and McConkey, 2003; Yang et al., 2004). In addition, bortezomib was shown to inhibit activation of caveolin-1, which is required for cell motility or migration (Podar et al., 2004).
19
b. Advantages of bortezomib as an anti-cancer agent
Bortezomib has several advantages for treatment of cancer. First, malignant cells are sensitive to treatment with proteasome inhibitors than normal cells (Chen et al., 2011a). It has been demonstrated that malignant cells habor the elevated activity and expression of proteasome, compared with normal cells (Kumatori et al., 1990; Arlt et al., 2009; Ma et al., 2009), suggesting the possibility that cancer cells are more dependent on proteasome activity for survival in diverse milieu. This hypothesis is supported by many preclinical studies (Soligo et al., 2001; Ovaa et al., 2004; Chen et al., 2005; Bazzaro et al., 2006; Chen et al., 2006). In addition, Hideshima et al. show that patient-derived myeloma cells are at least 170-fold more sensitive to bortezomib compared with peripheral blood mononuclear cells (PBMCs) from normal volunteers (Hideshima et al., 2001). Second, bortezomib can increase radiation sensitivity in cancer cells in vitro and in vivo (Pervan et al., 2001; Russo et al., 2001; Edelman, 2005; Goktas et al., 2010). Finally, bortezomib can overcome the resistance of conventional chemotherapeutic agents by enhancing their sensitivity (Schwartz and Davidson, 2004; Landis-Piwowar et al., 2006).
20 c. Combinatorial therapies
Although bortezomib has achieved significant benefit for multiple myeloma patients in the clinical trials, its effectiveness and administration have been limited by severe side effects, the acquisition of drug-resistance in a large portion of cancer patients, interactions with some natural products and decreased efficacy toward solid tumors (Chen et al., 2011a). According to many clinical studies, bortezomib possesses chemo-/radio-sensitizing activities as a means for overcoming resistance when combined with conventional therapeutic agents or radiation (Dou and Goldfarb, 2002; Kane et al., 2006). In addition, combined treatment with bortezomib and novel targeted therapies such as histone deacetylase (HDAC) inhibitors (vorinostat, belinostat, panobinostat and romidepsin) (McConkey and Zhu, 2008; Zhu et al., 2010), HSP90 inhibitors (tanespimycin, AUY-022) (Mitsiades et al., 2002; Roue et al., 2011) and other classes of targeted inhibitors (e.g. Akt inhibitors, pan-Bcl inhibitor, farnesyl transferase inhibitor, etc) have more clinical benefits as compared to bortezomib alone in various cancers. Overall, the combinatorial therapies have shown very promising results, and the most successful combination is likely to be approved soon to treat cancer patients.
21 F. Chloroquine
Chloroquine has long been used for treatment or prevention of malaria (https://en.wikipedia.org/wiki/Chloroquine). Although chloroquine was initially ignored for a decade because it was considered too toxic to use in humans, currently, chloroquine and its analogues receive attention for the treatment of dermatological, immunological, cancerous and infectious diseases (Mushtaque and Shahjahan, 2015). Particularly, oncologists are focusing the use of chloroquine on the treatment of highly aggressive and metastatic cancers, because it inhibits autophagy and sensitizes to radiation and chemotherapy (Kangwan et al., 2014; Vlahopoulos et al., 2014).
Chloroquine is a representative lysosomotropic agent (Moczar, 1981). Lysosomotropic property (Lysosomotropism) means the accumulation of weak bases in the acidic cellular organelles, such as lysosomes and endosomes. Chloroquine is a diprotic weak base, having charged (protonated) or uncharged (unprotonated) form. When chloroquine exists as the uncharged form (unprotonated form), it can freely and rapidly diffuses across the cellular membranes of the cell and organelles. When chloroquine enters into acidic organelles such as lysosome, it is charged (protonated form) and, thus, trapped in the organelles (Volkl et al., 1993; Borgonovo et al., 2002; Zhao et al., 2005). The accumulation of chloroquine in lysosome raises lysosomal pH and subsequently prevents proteolytic processes via the inhibition of the lysosomal enzyme activities (Law et al., 1984; Hostetler et al., 1985; Yin et al., 2003), consequently, chloroquine inhibits the latter step of autophagy process. Because autophagy seems to contribute to promote cancer, chloroquine may sensitize cancer cells through inhibiting autophagy (Kimura et al., 2013). The lysosomotropism of chloroquine is the most important feature for treatment of malignant cancer. Accumulation of chloroquine in lysosomes also causes cellular damage through the increase in the lysosomal volume and permeability
22
(Al-Bari, 2015). In addition, other mechanism of chloroquine for its anti-cancer effects include the inhibition of lysosomal enzymes, particularly phospholipase A2 (Nosal and Jancinova, 2002), the molecular intercalation into DNA (Gurova, 2009) and the suppression of the ATP-binding cassette (ABC), which plays an important role in drug transportation (Vezmar and Georges, 2000).
Among numerous sensitizers that have been examined, chloroquine appears to be particularly promising since it does not cause significant side effects, and can effectively sensitize cell killing effect by genotoxic agents in a cancer-specific manner (Solomon and Lee, 2009). Since extracellular environment of solid tumors is usually acidic than normal tissues (Vaupel et al., 1989; Boyer and Tannock, 1992; Newell et al., 1993), a combination of two drugs (i.e. weak DNA intercalating chloroquine and etoposide) can be selectively cytotoxic to the cells in acidic environment while cells in a normal microenvironment are protected (Jensen et al., 1994). Clinicaltrials.gov search identified 17 currently ongoing clinical studies evaluating the potential of chloroquine as a sensitizing agent and these Phase I/II studies demonstrate early evidence for application in anti-cancer therapy (Duffy et al., 2015).
23 G. Purpose of the study
Malignant gliomas, among which glioblastomas constitute the largest group, are characterized by a dramatically diffuse infiltration into the brain parenchyma and they are resistant to radiotherapy and conventional chemotherapy. Therefore, no patient with glioblastoma multiform (GBM) has been cured to date.
Although bortezomib is an attractive agent to treat malignant hematopoietic cancers (Hideshima et al., 2001), solid tumors are often resistant to bortezomib. Therefore, identification of the sensitizing agents that can overcome bortezomib resistance may provide an effective treatment strategy for malignant gliomas. In addition, to hinder the production of proteins responsible for the malignant behavior of GBM, chloroquine may be an effective drug and administration of chloroquine to inhibit autophagy in combination with anti-cancer therapies is currently evaluated in clinical trials.
In this study, we show that salubrinal effectively overcomes the resistance of malignant glioma cells to bortezomib, via eIF2α phosphorylation dependent- and independent manner. In addition, we showed that combined treatment with kaempferol and chloroquine effectively induces cell death in glioma cells via lysosomal rupture due to overloading of its cargos. Taken together, these results suggest that perturbation of preteostasis by aggravating the activites of proteasome and lysosome may provide potential therapeutic strategy for targeting malignant glioma cells.
24
II. MATERIALS AND METHODS
A. MATERIALS a. Chemicals
Bortezomib (Santa Cruz Biotechnology); 3-methyladenine (3-MA), bafilomycin A1 (BFA), crystal violet, MG132, DAPI, cychloheximide (CHX), chloroquine (CQ), hydroxychloroquine (HCQ), mefloquine, quinacrine, kaempferol, acridine orange (AO), monodansylacadeverine (MDC), sodium phenylbutyrate (4-PBA), monensin and sodium tauroursodeoxycholate (TUDCA) (Sigma Chemical Corporation); salubrinal, wortmannin, LY294002 and necrostatin-1 (Nec-1) (Selleckchem); benzyloxy-carbonyl-Val-Ala-Asp-(Ome)-fluoromethylketone (z-VAD-fmk) (R&D systems); calcein actoxymethyl ester (calcein-AM), ethidium homodimer-1 (EthD-1), LysoTracker® Red DND-99 (LTR), MitoSOXTMRed, BODIPYFL-pepstatin, HCS LipidTOX Deep Red and pHrodo® Green AM
Intracellular pH Indicator (Invitrogen); Proteostat® Aggresome detection kit (Enzo).
b. Antibodies
caspase-3 (Stressgen); caspase-8, caspase-9, CHOP, GRP78, eIF2α, anti-eIF2α, p70S6kinase, anti-p70S6kinase, anti-phospho-ribosomal S6, anti-anti-phospho-ribosomal S6, anti-cleaved PARP and anti-LC3B (Cell Signaling Technology); PARP, LAMP2 and gamma-H2AX (Abcam); α-tubulin, anti-CREB2 (ATF4), anti-Ub, anti-LAMP2 (Santa Cruz Biotechnology); anti-PDI (Enzo); anti-p62 (Sigma Chemical Corporation); anti- DR5 (Koma Bio); anti-p62 and anti-Cathepsin D (CD) (BD Biosciences Pharmingen); horseradish peroxidase-conjugated anti-rabbit IgG and horseradish peroxidase-conjugated anti-mouse IgG HRP (Invitrogen).
25 B. METHODS
a. Culture of glioma cell lines and normal human astrocytes
The human malignant glioma cell lines U251MG, T98G, and U87MG cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (GIBCO BRL, Life Technologies, Grand Island, NY) supplemented with 10% fetal bovine serum (FBS) and antibiotics (GIBCO-BRL, Life Technologies). The cells were incubated in 5% CO2 at 37℃. The primary cultures of
normal human astrocytes were prepared from 14-week gestation of fetal cerebrum tissues as described previously.15 Human astrocyte cultures were grown in DMEM with high glucose supplemented with 10% FBS and 20 mg/mL gentamicin, sub-cultured every 2weeks, and cell culture passage number of<5 were used in the present study.
b. Determination of cellular viability using calcein-AM and EthD-1 (Live-Dead assay)
Cells were cultured in 24-well plates and treated as indicated. For measurement of cellular viability, 2 μM calcein-AM, a green fluorescent indicator of the intracellular esterase activity of cells (live), and 4 μM EthD-1, a red fluorescent indicator of membrane-damaged (dead), were added to each well, and the plates were incubated for 5 min in 5% CO2 at 37℃.
Cells were then observed under a fluorescence microscope (Axiovert 200; Cal Zeiss) equipped with Zeiss filter sets #46 and #64HE. Viable cells, corresponding to those that exclusively exhibited green fluorescence, were counted in five fields per well at 200 x magnification. Only exclusively green cells were counted as live because bicolored (green and red) cells cannot be unambiguously assigned to live of dead groups. The percentage of live cells (Live %), calculated as green cells (green + red + bicolored cells), was normalized to that of untreated control cells (100%).
26
c. Determination of cellular viability by an MTT assay
Cells were plated in 96-well plates at a concentration of 1x104 cells/ml. After treatments, MTT assay was performed according to the manufacturer’s protocol (Sigma). Absorption at 570 nm was normalized to that of untreated control (100%), and the results were expressed as viability % of control.
d. Determination of synergism by the combination index (CI) method
The interaction of salubrinal and bortezomib/MG132 was determined by CI methods the CompuSyn software program (ComboSyn Inc., Paramus, NJ, USA) to determine whether the combination was additive or synergistic. Data from cell viability assays (Live-Dead assay) were expressed as the fraction of anti-proliferation activity (cell viability loss) by the individual drugs or the combination in drug-treated cells, and CI values between two drugs were generated. A CI of 1 indicates an additive effect, whereas a CI of <1 indicates synergy. A CI value at 50% effect (Fa=0.5) based on the CI isobologram equation was also calculated (Chou and Talalay, 1984).
e. Colonogenic cell survival assay
T98G cells were plated in 12-well plates at a density of 1ⅹ103 cells per well in triplicate and treated with salubrinal alone, bortezomib alone, and combination for 8 h. U251MG cells were also plate in 12-well plates at a density of 1ⅹ103 cells per well in triplicate and treated with kaempferol alone, chloroquine alone, and combination for 12 h. Then, the cells were treated with fresh drug-free medium and incubated for an additional 10 days. Cells were fixed in cold-methanol, and stained with 0.5% crystal violet. Image acquisition was conducted using a digital camera.
27 f. Western blotting
Cell were washed in PBS and lysed in boiling sodium dodecyl sulfate/polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer (62.5 mM/L Tris [pH6.8], 1% SDS, 10% glycerol, and 5% β-mercaptoethanol). The lysates were boiled for 5 min, separated by SDS-PAGE, and transfected to an Immobilon membrane (Millipore). After blocking nonspecific binding sites for 30 min by 5% skim milk, membranes were incubated for 2 h with specific Antibodies. Membranes were then washed three times with TNET buffer (50 mM Tris-HCl [pH7.4], 150 mM NaCl, 5 mM EDTA, 0.05% Tween20) and incubated further for 1 h with horseradish peroxidase-conjugated anti-rabbit or mouse. Visualization of protein bands was accomplished using ECL (Advansta).
g. Establishment of the stable cell lines expressing the fluorescence specifically in endoplasmic reticulum
To establish the stable cell lines expressing the fluorescence specifically in endoplasmic reticulum (ER), T98G cells were transfected with the pEYFP-ER vector (Clontech). The
pEYFP-ER vector encodes a fusion protein consisting of EYFP, flanked on the 5′-end by the
ER-targeting sequence of the luminal-resident protein calreticulin (Fliegel et al., 1989) (Fliegel, l, et al., (1989) J boil Chem 264:21522-21528), and on the 3′-end by a conserved KDEL motif present in luminal ER proteins (Munro and Pelham, 1987; Pelham, 1996). Therefore, the fluorescence derived from the pEYFP-ER vector is expressed in the ER lumen. Stable cell lines expressing pEYFP-ER (YFP-ER) were selected with fresh medium containing 500 μg/mL G418 (Calbiochem).
28 h. Immunocytochemistry
Cells were fixed in methanol/actone (1:1) for 5 min at -20℃ and washed in PBS. Fixed cells blocked in 5% BSA plus 0.1 M glycine for 45 min. Primary antibodies [anti-Ub (1:200, mouse) and anti-LAMP2 (1:200, goat) (Santa Cruz Biotechnology); anti-p62 (1:1000, rabbti) (Sigma Chemical Corporation); anti-LC3B (1:200, rabbit) (Cell Signaling Technology); anti- anti-Cathepsin D (1:200, rabbit) and anti-pre-Cathepsin D (1:200, rabbit) (Invitrogen); anti-γ-H2AX (Abcam)] were incubated o/n in block at room temperature and then washed three times in PBS. Anti-mouse Alexa Fluor 488 (1:1000, Molecular Probes) was incubated for 45 min at room temp. Slides were mounted with ProLong Gold antifade mounting reagent (Molecular probes) and visualized with a fluorescence microscope using Zeiss filter sets #1, #20 and #46HE.
i. Small interfering RNAs
The 25-nucleotide small interfering RNA (siRNA) duplexes purchased from Invitrogen and their sequences are as follows: DR5. BLOCK-iTTM Fluorescent Oligo (Invitrogen) of Negative Universal ControlTM (Invitrogen) was used as the control. After annealing of the pairs of siRNA oligos, Cells were transfected with siRNA oligonucleotides using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s recommendations. To estimate the transfection efficiencies in the various siRNA experiments using siRNAs, we transfected cells with a fluorescently labeld control oligonucleotide (BLOCK-iTTM Fluorescnet Oligo) and assessed the percentage of green fluorescent cells via fluorescence microscopy. In all of our transfection experiments using siRNAs, the estimated transfection efficiencies (i.e., the percentages of green fluorescent cells) were greater than 95%. To confire successful siRNA-mediated knockdown, we performed Western blotting of the proteins of interest.
29
j. Knockdown experiments using small hairpin (sh) RNA
For the knockdown experiments employing Cathpesin D-targeting shRNA, HEK293TN cells were transfected with the plasmid containing the non-targeting shRNA (SHC002V, Sigma-Aldrich) or the plasmid containing Cathepsin D-targeting shRNA (TRCN0000003660, TRCN0000003661, Sigma-Aldrich), together with pMD2.G (the envelope plasmid) and pPsAX2.0 plasmid (the packaging plasmid), using TransIT-2020 transfection reagents (Mirus Bio LLC) according to the manufacturer’s instructions. After 48 h of lentiviral particle production, U251MG cells were infected with the filtered lentiviral medium (derive from HEK293TN cultures) supplemented with 4 μg/ml polybrene.
k. Assay of aggresome formation
Cells were plated on cover slips and treated. The cells were washed with PBS, and fixed in 4% formaldehyde in PBS for 30 min at room temperature. Then the cells were permeabilized with 0.5% Triton X-100 in PBS, and ProteoStat® dye (ProteoStat® Aggresome detection Kit, Enzo Life Sciences) was added. The samples were incubated for 30 min at room temperature. The cells were washed with PBS, covered with slide glasses and observed with a fluorescence microscope using Zeiss filter sets #64HE.
l. Visualization of autophagic vacuoles
Following treatment with kaempferol, autophagic vacuoles were labeled with MDC by incubating cells frown on coverslips with at 50 μM MDC in PBS at 37℃ for 15 min. After incubation, cells were washed three times with PBS and immediately analyzed by fluorescence microscopy using an inverted microscope (Axiovert 200, Zeiss) equipped with Zeiss filter set #34.
30 m. LysoTracker Red staining
To detect lysosomes, cells treated with kaempferol. Treated cells were stained with LysoTracker® Red DND-99 at 100 nM for 20 min and were observed with a fluorescence microscope using Zeiss filter sets #64HE.
n. Detection of acidic vesicular organelles (AVOs) with acridine orange staining
To detect AVOs, cells treated with kaempferol and/or chloroquine were stained with 1 μg/ml acridine orange for 15 min and samples were observed under a fluorescence microscope using Zeiss filter set #64HE.
o. BODIPY FL-pepstatin staining
The catalytic activity of cathepsin D declines essentially to zero on going from pH3.0 to pH 7.0, and we suggest that the binding site for substrate and pepstatin is abolished by a conformational change in the enzyme molecule (Knight and Barrett, 1976). To stain the fixed cells with BODIPY-FL-pepstatin A, U251MG cells were fixed and permeabilized simultaneously in 3% formaldehyde and 0.1% glutaraldehyde in sodium acetate buffer (pH 4.5). After several washes in the sodium actate buffer (pH 4.5), the cells were incubated in the same buffer containing 1 μM BODIPY- FL-pepstatin A at room temperature for 1 h. In some experiments, 500 μM of unlabeled pepstatin A was added in the BODIPY FL-pepstatin A solution to study the effect of competitive binding for Cathepsin D (Chen et al., 2000).
p. HCS LipidTOXTM neutral lipid staining
Cells were plated on cover slips and treated with kaempferol and/or chloroquine. The cells were washed with PBS, and fixed in 4% formaldehyde in PBS for 20 min at room
31
temperature. Fixed cells were stained with HCS LipidTOXTM Red neutral lipid stain (Molecular ProbesTM, 1:1000 in PBS) for 30 min and were observed with a fluorescence microscope using Zeiss filter sets #64HE.
q. Live-cell FluoZin-3 experiments
Cultured cells were washed twice with Live Cell Imaging Solution (Molecular Probes, 140 mM NaCl, 2.5 mM KCl, 1.8 mM CaCl2, 1.0 mM MgCl2, 20 mM HEPES, pH=7.4) before
being incubated in Live Cell Imaging Solution supplemented with 10 μM FluoZin-3 for 25 min at 37℃ (in the dark). Stained cells were washed twice with FluoZin-3-free live cell imaging solution and were observed with a fluorescence microscope using Zeiss filter sets #20.
r. Intracellular pH detection
Cells were plated on cover slips and treated with kaempferol and/or chloroquine. Cells were washed with Live Cell Imaging Solution and stained in mixed solution with pHrodoTM Red AM (Molecular Probes, 1:1000), Power LoadTM (Molecular Probes, 1:100) and Live Cell Imagine Solution (Molecular Probes) for 30 min at 37℃. Stained cells were washed with Live Cell Imaging Solution and observed with a fluorescence microscope using Zeiss filter sets #20.
s. Transmission electron microscopy
Cells were prefixed in Karnovsky’s solution (1% paraformaldehyde, 2% glutaraldehyde, 2 mM calcium chloride, 0.1 M cacodylate buffer, pH 7.4) for 2 h and washed with cacodylate buffer. Post-fixing was carried out in 1% osmium tetroxide and 1.5% potassium ferrocyanide for 1 h. After dehydration with 50-100% alcohol, the cells were embedded in Poly/Bed 812 resin (Pelco, Redding, CA) and polymerized, and observed under electron microscope (EM
32
902A, Zeiss, Oberkonhen, Germany).
t. Measurement of mitochondrial superoxide production
To measure mitochondrial superoxide production, cells were loaded with 2.5 μM MitoSOX-Red for 20 min, washed with Hank’s Buffered Salt Solution (HBSS) with Ca2+ and Mg2+, and further processed for flow cytometry.
u. Statistical analysis
All values were presented as mean ±S.D. (standard deviation) from at least three separate experiments. Statistical significances of differences were determined using Student’s t-test or one-way ANOVA followed by a Bonferroni multiple comparison tests, as indicated in the figure legend. A P-value < 0.05 was considered significant.
33 PART1
eIF2α modulates bortezomib-induced vacuolation and cell death in glioma cells via phosphorylation-dependent and independent manner
34 1. Introduction
[Glioma]
Glioma is a common type of primary brain tumor and almost 80% of malignant brain tumors corresponds to gliomas (Dolecek et al., 2012). While many of benign brain tumors are gliomas, almost 80% of malignant brain tumors are gliomas. Despite the development of diagnosis and surgical techniques, malignant gliomas are still associated with high morbidity and mortality (Wen and Kesari, 2008). In addition, cytotoxic agents, e.g. temozolomide (TMZ), have limited efficacy in recurrent disease (Wong et al., 1999a; Wong et al., 1999b). Therefore, there is an urgent need for new and improved treatment strategies against gliomas.
[Glioma & UPS]
The ubiquitin-proteasome system (UPS) undergoes a complex mechanism where proteins are first targeted for degradation by the ubiquitination machinery and then recognized, unfolded and proteolyzed by the proteasome (Finley, 2009). Proteins targeted by UPS include a broad array of regulatory proteins that play important roles in cell cycle progression, cell development and differentiation, DNA damage responses, and tumorigenesis. Dysfunction of UPS gives rise to various kinds of diseases including infectious diseases, autoimmune diseases and cancers (Du and Mei, 2013). Actually, elevated proteasomal activity has been detected in many types of cancers (Ren et al., 2000; Wyke et al., 2004; Chen and Madura, 2005; Arlt et al., 2009). Also, proteasome activity was elevated in both primary and recurrent glioblastoma compared to normal brain tissue (poster, targeting proteasome activity with marizomib as a therapeutic perspective for glioma patients, Shahrzad Jalali et al. SNO-triphase-poster 16Nov2015). Accumulating evidence has shown the dysregulation of UPS components in
35
cancer and increased stabilization of oncoproteins as well as destabilization and degradation of tumor-suppressor proteins is considered to be important for brain tumor development. Therefore, UPS is an unquestionable therapeutic target for brain tumor treatment (Hede et al., 2014).
[Proteasome inhibitor & solid tumor]
Proteasome inhibition demonstrates anticancer activity through various mechanisms, including induction of apoptosis, disruption of cell cycle progression, inhibition of proliferation, and anti-angiogenesis (Boccadoro et al., 2005). The successful development of the proteasome inhibitor bortezomib as an anticancer drug has improved survival in patients with multiple myeloma. Bortezomib is active against a wide variety of tumor cell types such as non-Hodgkin’s lymphoma, mantle cell lymphoma and non-small cell lung cancer in vitro (Orlowski et al., 1998; Fanucchi et al., 2006; Kane et al., 2007). However, as a monotherapy, bortezomib has demonstrated poor responses in both phase I and II trials of a broad number of solid tumors (Resistance to proteasome inhibitors in cancer, Q.Ping Dou, 9.3. Preclinical studies to demonstrate the efficacy of proteasome inhibitors in solid tumors cells, p.238). One of approaches for overcoming limitation of bortezomib in solid tumor is the combination treatment with other conventional chemotherapy (Johnson, 2015). Actually, Phase II study of bortezomib in combination with gemcitabine/carboplatin was successfully conducted in chemotherapy-naïve advanced non-small cell lung cancer (NSCLC) patients (Davies et al., 2009), suggesting that bortezomib-based combinations could be proven promising for solid tumor.
36
The ER is a dynamic and essential organelle for the synthesis, assembly, and post-translational modification of surface and secretory proteins. In addition, the ER is responsible for lipid synthesis and regulation of intracellular calcium levels (English and Voeltz, 2013). The ER is an interconnected system of sheets and tubules (Shibata et al., 2009). In all eukaryotes the ER network is extremely dynamic with tubules undergoing continuous fusion and fission reactions (Ajitkumar et al., 1988; Prinz et al., 2000). Fragmentation of the ER and the Golgi apparatus has been extensively described in dying cells, in addition to alterations in the morphology of the mitochondria (Urra and Hetz, 2012). ER swelling is a morphological change associated with multiple cell death modalities (Van Cruchten and Van Den Broeck, 2002). Swelling and dilation of the ER is a hallmark of chronic ER stress (Encyclopedia of cell biology, 1st edition, Ralph Bradshaw). Although it is becoming clear that many stress conditions induce drastic ER-morphological changes, the contribution of this process to apoptosis remains largely elusive (Urra and Hetz, 2012). Disturbances in any of the vital functions results in an increase of unfolded proteins in the ER, which initiate the phenomenon known as endoplasmic reticulum stress response (ERSR) (Schroder and Kaufman, 2005). The goal of the ERSR is to enhance protein folding capabilities, reduce new protein synthesis, and clear malformed proteins to allow the return of normal cellular function (Johnson et al., 2011). If the ER stress is prolonged, apoptotic pathways can be triggered by activation human caspase-4/murine caspase-12 that localizes to the ER membrane (Hitomi et al., 2004). Proteasome inhibitors induce ER stress that means the accumulating condition of misfolded or unfolded proteins in the ER (Obeng et al., 2006) and activate the unfolded protein response (UPR), an adaptive program to restore proteostasis (Walter and Ron, 2011).