생명 다양성 보전에 활용되는 보전유전학 연구동향 전형배 Page 1 / 38 BRIC View 2020-T25
생명 다양성 보전에 활용되는 보전유전학 연구동향
전 형 배
Concordia University, Montreal E-mail: jhb5696@gmail.com 요약문 인간에 의한 대량 멸종은 생명 다양성의 위기를 가속화시키고 있고, 이는 인류의 존립을 위태롭게 하므로 반드시 극복되어야 한다. 보전유전학은 다음과 같은 다양한 방법을 통해 생명 다양성의 유지에 기여한다. 첫째, DNA 바코딩과 이를 응용한 eDNA와 같은 세밀한 진 단 도구를 통해 생명 다양성의 현황을 진단할 수 있게 한다. 둘째, 분자 계통학적 방법론은 기존에 밝혀지지 않았던 은밀 다양성을 발굴하는데 기여하고 생명 다양성의 생성 메커니즘 인 종 분화를 이해하는데 기여할 뿐만 아니라 보전이 필요한 생물들의 실체를 밝히는데 기 여한다. 셋째, 생명 다양성을 위협하는 요인들에 대해 진단하는 다양한 도구를 제공해주어 보다 효과적인 보전 전략의 수립을 가능하게 한다. 이처럼 보전유전학은 집단의 보전을 위 한 다양한 학제의 연구들을 통해 집단의 절멸을 야기하는 원인을 파악하는데 도움을 줄 뿐 만 아니라, 생명 다양성의 생성 및 유지에 기여하는 메커니즘을 이해하는데 도움을 줌으로 써 효과적인 보전을 위한 근거 기반의 전략을 수립하는데 핵심적인 역할을 해왔다. 그 어느 때보다 대량 멸종의 위기에 직면한 현 시점에서 생명 다양성의 보전을 위한 학문 영역으로 써 보전유전학의 중요성은 날로 증대되고 있으며, 기존의 전통적인 생태학 영역에서 지녔던 한계를 보완하는 것을 넘어 점차적으로 대체해 나가고 있다. 여기에는 최근의 NGS 기술의 광범위한 보급도 한 몫하고 있다. 불과 수 년 전만 하더라도 유전체 정보는 인간과 모델생 물들의 전유물이었지만, 이제는 비단 인간뿐만 아니라 풍부한 유전체 수준의 분자 데이터를 거의 모든 생물분류군에 활용할 수 있는 시대가 열리고 있다. 이러한 변화는 보전유전학 분 야의 성장을 보다 가속화 할 것으로 전망된다. 이에 본 동향 리포트에서는 다양한 보전유전 학의 기법과 연구 동향을 소개하고자 하였다.
Key Words: Conservation Genetics, Population Genetics, eDNA, Biodiversity, Phylogenetics
생명 다양성 보전에 활용되는 보전유전학 연구동향 전형배 Page 2 / 38
목 차
1. 서론 – 멸종의 메커니즘과 보전유전학 2. 본론 2.1. 진단: 생명 다양성의 평가 2.1.1. DNA바코딩과 eDNA를 통한 생명 다양성 평가 2.1.2. 분자계통학을 통한 생명 다양성의 발굴2.1.3. 보전 단위(Conservation units)와 보전의 우선순위(Conservation priority) 선정 2.2. 유전적 다양성의 평가
2.2.1. 중립 변이
2.2.1.1. 유효 집단 크기(Effective population size)
2.2.1.2. 유효 집단 크기의 역사적 변동(Demographic history) 2.2.1.3. 집단의 유전적 구조(Population structure) 2.2.1.4. 과거에 분화된 분류군 사이의 유전적 흐름 2.2.1.5. 가계분석 2.2.2. 적응 변이 2.2.2.1. 후보 유전자(Candidate genes)
2.2.2.2. 유해 변이의 축적(Accumulation of Deleterious mutation) 2.2.2.3. 지역 적응(Local adaptation) 2.3. 집단의 유전적 다양성이 유지되는 메커니즘 2.4. 인간 활동에 의해 야기되는 절멸과 진화적 압력(Human-induced evolution) 2.4.1. 포획에 의한 진화(Harvesting-induced evolution) 2.4.2. 인위적 서식지 변화와 그에 따른 진화 2.4.3. 생물의 인위적 이주에 의한 진화 2.5. 보전유전학이 제안하는 보다 적극적인 보전 전략과 그 유의점 3. 결론 4. 참고문헌
1. 서론 – 멸종의 메커니즘과 보전유전학
인간은 태초부터 자신을 둘러싼 생물들이 사라지는 것을 두려워 했고, 그 두려움의 흔적은 노아의 방주와 같은 전설과 신앙 속에서 전승되어 오고 있다. “왜 인간은 생물들의 손실을 두려워 하는 걸까?” 해답은 단순하다. 그들로부터 우리의 생존을 위한 모든 것을 얻기 때문이다.생명 다양성 보전에 활용되는 보전유전학 연구동향 전형배 Page 3 / 38 이러한 생명 다양성의 중요성에도 불구하고, 생명 다양성은 급속도로 감소되어가고 있다. 그리고 그것을 부정할 수 있는 증거는 현재로서는 빈약하다. 대량 멸종에 대한 우려는 일부 전문가의 우려에 그치지 않고 이제 사회 전반적으로 확대되어가고 있다. 하지만, 이러한 우려와 관심에도 불구하고 생명 다양성의 감소에 적절히 대처하기 위한 대책 마련은 여전히 더딘 상황이고, 심지어 그 대책이라는 것도 어떤 근거를 기반으로 한 것인지 불분명한 것들이 대다수인 상황이다. 보전유전학은 그동안 누적된 집단 유전학(population genetics) 기반의 이론들을 통해 멸종이 일어나는 원리를 밝히고 멸종을 방지하고 생명 다양성을 북돋아 주기 위한 근거를 제공해줄 수 있는 학문 분과이다 [1, 2]. 그림 1. Extinction vortex [1, 2]. 보전유전학에서 말하는 멸종이 일어나는 핵심적인 원리는 ‘절멸 소용돌이(extinction vor-tex)’로 요약된다. 자연재해, 인위적 교란, 병원체 등의 다양한 원인에 의해 집단의 크기가 감소되면, 다음 세대에 대물림해 줄 수 있는 유전적 잠재력이 감소된다. 한 집단에서 다음 세대의 유전적 다양성에 기여하는 개체의 숫자를 ‘유효 집단 크기(effective population size)’라 하며 [3], 감소된 유효 집단 크기는 생존에 관련된 기능적 다양성을 단순화시킬 개연성을 높인다. 단순화된 기능과 형질은 시시각각 변화하는 외부환경에 임기응변하는 능력을 소실시키고, 이로 인해 집단을 구성하는 개체 수는 또 다시 감소하게 된다. 이는 직접적으로 다음 세대에 더욱 작은 유효 집단 크기를 대물림하는 결과를 초래한다. 이는 또 다른 문제를 야기하는데 우연히 만들어진 유해한 돌연변이들이 높아진 근친 교배(inbreeding)의 위험으로 인해 단 몇 세대 만에 집단에 급격히 확산될 수 있다.
생명 다양성 보전에 활용되는 보전유전학 연구동향 전형배 Page 4 / 38 이처럼 유효 집단 크기의 여러 세대에 걸친 감소는 궁극적으로 절멸의 소용돌이를 야기한다 [4]. 마치 나비 한 마리의 날갯짓으로 태풍이 만들어지는 것에 비견할 수 있다. 보전유전학은 절멸 소용돌이에 영향을 주는 여러 요소들을 진단하고, 부정적 효과를 갖는 요소들을 배제하기 위한 중요한 이론적 근거를 제공하는데 그 목표가 있다. 본 동향리포트에서는 그동안 보전유전학이 생명 다양성 보전을 위해 기여해온 연구 주제들을 소개하고 앞으로의 전망에 대해 간략히 살펴보고자 한다. 일부 내용들은 기존의 브릭에 연재했던 <분자생태학 톺아보기>(링크: https://www.ibric.org/myboard/list.php?Board=news& PARA3=49)를 인용했음을 밝힌다.
2. 본론
2.1. 진단 : 생명 다양성의 평가
2.1.1. DNA 바코딩과 eDNA를 통한 생명 다양성 평가 생명 다양성을 보전함에 있어 보전 대상의 존재 유무를 인지하는 것은 보전의 첫 단추이다. 황새를 보전한다면서 백로와 황새를 구분할 수 없다면 어떻게 보호할 수 있을까? 종을 구분할 수 있는 능력(동정, identification)을 일부 사람만 갖고 있거나, 그 구분 방법이 불완전하다면, 효과적인 종 보전은 불가능할 것이다. 종을 구분하는 동정 능력은 해당 분류군의 형태적 차이가 미미하거나, 형태적 특징이 손상되기 쉬워 식별하기 어렵거나, 동정 방법이 너무 전문적이어서 일반인 수준에서 도저히 적용하기 어렵거나, 동정을 위해 생물의 살생이 불가피할 때 장벽에 부딪히게 된다. DNA 바코딩은 이런 한계를 극복할 수 있는 훌륭한 대안으로 제시되어 현재 광범위하게 활용되고 있다 [5]. DNA 바코딩이란 쉽게 말해 유전자 단편만으로 생물종의 이름을 알아내는 기술을 말한다 [5]. DNA 바코딩은 같은 종 내에서는 유사성을 갖지만, 다른 종 사이에는 차이를 갖는다는 유전자가 존재한다는 가정에서 출발한다. 만약 어떤 유전자 단편에서 종간 변이는 5~20%, 종내 변이는 0~2% 범위에 있다는 사실이 확인되었다면, 해당 유전자 단편을 통해 종을 동정할 수 있다. 지구상에 존재하는 생명다양성의 DNA 라이브러리를 적절히 구축해두기만 한다면, 누구나 손쉽게 보전이 필요한 생물들의 이름을 알아낼 수 있는 것이다. 효과적인 바코드 유전자가 가져야 할 조건은 크게 두 가지가 알려져 있다. 우선 바코드 유전자들은 종 간에는 충분히 다르고, 종 내에서는 충분히 유사해야 한다. 이를 달리 말하면 유전적 거리(genetic distances)가 종 간에는 멀고 종 내에는 가깝다는 것이다. 유전적 거리를 기반으로 바코드 유전자의 효용성을 판별하기 위해서는 바코딩 갭(barcoding gap)의 존재 여부를 확인한다 [6, 7]. 여러 지역의 종내 종간 표본들로부터 바코드 유전자 서열을 확보한 뒤 각각의 염기서열 사이의 유전적 거리를 계산한다. 그리고 그 유전적 거리의 빈도 분포표를 통해 종간 종내 유전적 거리의 빈도가 중첩되지 않는 격차(gap)의 유무를 확인하게 되는데, 이 격차가 나타나는 구간을 “바코딩 갭”이라 한다. 만약 어떤 유전자에서 종내 유전적 거리와 종간 유전적 거리의 빈도 분포 사이에생명 다양성 보전에 활용되는 보전유전학 연구동향 전형배 Page 5 / 38 갭이 존재한다면, 효과적인 바코드 유전자로 사용될 수 있다 [8]. 간혹 일부 분류군에서 바코딩 갭이 불분명한 경우가 존재하는데, 이 경우는 크게 두 가지로 일부 표본에서 종간 유전적 거리가 낮거나, 종내 유전적 거리가 큰 경우가 그에 해당한다. 종간 유전적 거리가 가까운 경우는 다른 종으로 분류된 경우가 실제로는 가까운 종인 경우이거나, 종간 교잡에 의해 다른 종의 변이가 도입된 결과일 수 있다. 특히 분류학적인 충분한 연구가 이루어지지 않은 상태의 분류군에서는 같은 종에 대해 다른 종으로 분류한 동종이명(synonym)의 경우가 흔히 관측된다. 한편, 종내 유전적 거리가 큰 경우는 실제 다른 종 수준으로 분화된 집단이지만, 아직 분류학적으로 기재가 되지 않은 경우가 일반적이며, 이 경우 추가적인 계통분류학적 연구를 통해 새로운 종을 발굴할 근거로 활용될 수 있다 [7]. 두 번째 조건은 하나 혹은 소수의 프라이머로 확인하고자 하는 분류군의 모든 종이 증폭되어야 한다는 것이다. 다시 말해, 수많은 종들을 증폭할 수 있는 universal primer를 활용할 수 있는 유전자여야 한다 [9]. 따라서, 프라이머가 결합되는 가장자리 부위(flanking regions)는 여러 종 사이에 변이가 적은 보존적인 특성을 지니고 있어야 한다.
위의 기준을 잘 충족하는 DNA 바코드로 활용되는 유전자로는 mitochondrial cytochrome c oxidase subunit I (COI)가 동물에서 알려져 있다. COI 유전자 내에서 특정한 500~600 bp 구간은 양 말단에 여러 종에 걸쳐 보존적인 구간이 존재하여 여러 종을 아우르는 universal primer를 디자인할 수 있고, 그 중간에는 종에 따른 변이가 존재하는 구간이 존재하므로, 이는 바코드 유전자가 가져야 할 장점을 보여준다 [5]. 이 외에도 12S rRNA, 16S rRNA, cytochrome b (cyt b)가 사용되고 있다. 식물에서는 maturate K (matK), rbcLa, trnH-psbA intergenic spacer region이 이용된다.
DNA 바코딩은 신뢰할 수 있는 보편적인 종 구분 방법으로 점차 그 중요성이 증가되고 있다. DNA 바코딩 개념이 제안된 이후 지난 수십 년간 수 많은 분류군에서 DNA 바코드 라이브러리가 구축되어왔다. 머지않은 미래에 지구상 생명 다양성의 DNA 바코드 라이브러리가 완전히 구축된다면, 누구나 종 구분에 대한 훈련을 받지 않더라도 간단한 유전자 분석만으로 지구상 모든 종을 동정하는 것이 가능해질 것이다. 한편, DNA 바코딩은 일반적으로 동정을 하고자 하는 개체 하나하나를 개별적으로 비교해야 한다는 단점이 있다. 이 단점을 극복하기 위해 개발된 방법이 특정한 지역의 군집 전체를 통째로 시퀀싱하여 해당 지역 생물상을 규명하는 메타바코딩(metabarcoding)이다 [10]. 메타바코딩을 위한 시료들은 여러 방법을 통해 확보되는데, 이를 테면 미소생물의 경우 특정 지역의 군집 전체를 분쇄해서 DNA를 추출한 뒤 universal primers를 이용해 PCR하고 NGS 기반의 시퀀싱을 수행하여 종을 동정한다 [11]. 이를 위해서는 시퀀싱 플랫폼이 읽을 수 있는 리드 길이에 맞추어 프라이머를 디자인하게 된다 [12]. 최근에는 아예 환경에 존재하는 DNA 조각들을 직접적으로 시퀀싱하는 환경 DNA (environmental DNA) 기법이 활용되기 시작하고 있다 [13, 14]. 환경 DNA는 수중, 토양 그리고 대기 중에 존재하는 생명체에서 유래된 DNA와 그것을 분석하는 기술을 말한다. 초창기의 환경 DNA는 토양과 물에 사는 미생물들을 DNA를 통해 생물상을 파악하던 방식으로부터 유래했다. 환경 DNA는 높은 민감도, 시간과 비용의 절감, 안전성, 비파괴성, 높은 재현성 등의 장점을 갖고 있어서 향후 전통적인 생명 다양성 평가 방법을 대체할 것으로 기대되고 있다 [13]. 현재 환경 DNA 기법은 보전에 핵심적인 멸종 위기종 [15, 16] 혹은 보전에 위협 요인인 외래종의 서식 여부 탐지 [17, 18]와 개체군 크기의 추정 [19], 번식 생태에 대한 연구 [20] 뿐만 아니라 우리 생존에 중요한 자원으로 활용되는 생물들의 자원량 추정 [21]에도 적용되기 시작하고 있다.
생명 다양성 보전에 활용되는 보전유전학 연구동향 전형배 Page 6 / 38 이처럼 분자 생태학적 연구 기법인 DNA 바코딩과 메타바코딩 그리고 환경 DNA는 보전 대상이 되는 생물상을 신속하고 쉽고 정확하게 평가할 수 있게 하며, 이는 조사 풍경을 전반적으로 변화시켜왔고 앞으로도 그 발전 가능성이 높다고 볼 수 있다. 하지만, DNA 바코딩과 이를 응용한 환경 DNA의 높은 가치에도 불구하고 한계와 단점은 분명히 존재한다. 이를 극복하기 위해서는 다음의 사항이 반드시 고려되어야 한다. 기본적으로 DNA 바코딩은 내가 확인한 염기서열과 기존의 GenBank에 등록된 염기서열을 대조한 뒤 유사성을 기준으로 종을 동정하는 과정을 거치기 때문에 만약 오동정된 표본에서 추출된 염기서열이 GenBank 혹은 BOLD에 등록되어 있다면, 이후에 DNA 바코딩을 통해 얻어진
염기서열들은 모두 잘못된 결과를 보여줄 것이다. 따라서 공신력 있는 바코드 라이브러리(a reliable DNA barcode library)가 구축되어야 한다 [22].
현재로서는 오류가 있는 바코드 서열을 등록하는 연구자를 막을 방법은 없는 상황이다. 그리고 오류가 있는 서열을 공공데이터베이스에서 타인에 의한 삭제나 수정이 불가능한 상황이다. 그래서 연구자들은 각각 오류가 있는 서열을 상습적으로 등록하는 연구자들에 대한 개인적인 블랙리스트를 만들기도 한다. 하지만, 이는 매우 소모적인 과정을 동반하므로 이를 해결하기 위한 몇 가지 기준이 제시될 필요가 있다 [22]. 이를테면, 다음과 같은 바코드 서열 등록 과정은 오류가 있는 서열을 방지하는데 도움을 줄 수 있다. 1) 해당 분류군의 분류를 연구한 복수의 전문가가 참여하여 전통적 방법으로 종 동정을 실시하고, 2) 해당 시료를 보관과 열람이 용이한 곳에 보관한 뒤, 3) 그 시료들로부터 오염되지 않은 염기서열을 확보하고 4) 표본보관소 정보 및 표본 번호(voucher number)와 함께 해당 서열을 모두가 열람할 수 있는 GenBank, BOLD 등의 데이터베이스에 등록해야 할 것이다. 그리고 오류가 보고되었을 때에는 즉각적으로 오류에 해당되는 서열의 등록을 철회 또는 정정해야 한다 [22]. 2.1.2. 분자계통학을 통한 생명 다양성의 발굴 우리가 인지하고 알고 있는 생명 다양성을 보전하는 것 못지 않게 중요한 것은 미처 우리가 알지 못하는 생명 다양성을 발굴하는 것이다. 분자계통학(phylogenetics)은 생명 다양성을 발굴하는 중요한 이론적 근거들을 제공해주었다. 그 결과 지금까지 숨어있던 은밀종(cryptic species)들이 속속 발표되고 있다. 은밀종의 발굴은 보전 전략 측면에서 매우 중요한 의미를 갖는다. 일례로 국내에 서식하는 야생동식물을 보전하기 위한 ‘야생동식물보호법’ 상에서 보전의 단위는 종(species)이므로 법으로 규정된 보호 대상종은 보호를 받지만, 그 보호 대상이 아닌 종들은 보전의 수혜에서 제외된다. 따라서, 만약 하나의 종이 다양한 은밀종으로 구성되어 있음에도 발표되지 않는다면, 현행법(2020년 기준)상 이 은밀종을 보전할 법적 근거는 없는 셈이다. DNA 바코딩과 최신의 분자계통학적 연구들은 기존에 형태적으로 유사하여 같은 종으로 여겨져 왔던 종들이 이미 종 분화가 완성된 상호 독립적인 은밀종으로 구성되어 있음을 보여준다 [23]. 이처럼 은밀종의 발굴은 법적인 보전의 대상이 되지 않았던 종들에게 보전 정책의 대상이 될 수 있는 기회를 제공한다는 점에서 그 보전유전학적 의의가 있다 [24].
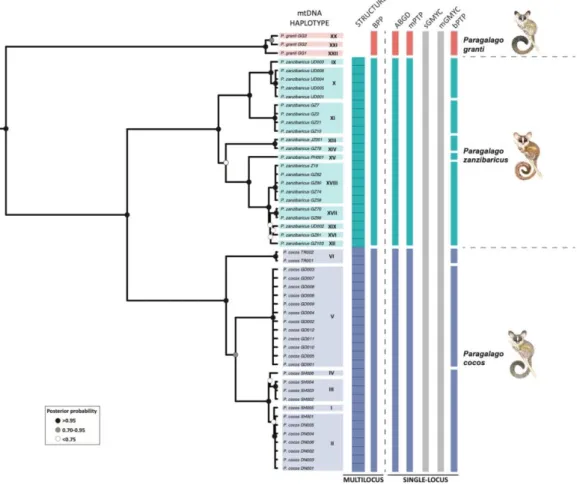
생명 다양성 보전에 활용되는 보전유전학 연구동향 전형배 Page 7 / 38 분자계통학을 통한 은밀종의 발굴은 크게 아래의 세 가지 과정으로 이루어진다 [25]. 첫째, 미세하게나마 형태적으로 구분되는가? 은밀종의 발굴에는 미세한 형태적 차이가 실마리로 활용되어 왔다. 은밀종 발굴을 위해 필수적으로 이용되는 분자 데이터를 생산하기 위해서는 비용이 발생하기 때문에 연구하고자 하는 분류군에 대한 아무런 확신이 없는 상태에서 분자 데이터를 취하기는 쉽지 않다. 이 단계는 최근 분석 비용이 낮아지고 전체 군집 수준의 분자 데이터를 생산하는 메타바코딩 기술이 보급됨에 따라 점차 생략되어가고 있다. 둘째, 유전적 분화가 존재하는가? DNA 바코딩을 통해 종내 유전적 거리가 먼 집단들을 확인함으로써 잠재적인 은밀종을 탐지할 수 있다. 만약 어느 집단 간 유전적 거리가 다른 종 사이의 유전적 거리에 준하는 수준이라면, 그 집단들은 서로 분화된 잠재적 은밀종일 수 있다. NGS 기반의 메타바코딩은 이 유전적 분화가 있는 집단들을 찾는데 효과적이다.
그림 2. Paragalago속의 원숭이의 종간 경계설정(species delimitation)의 사례.
생명 다양성 보전에 활용되는 보전유전학 연구동향 전형배 Page 8 / 38 셋째, 여러 유전자를 이용한 분자계통수 상에서 단계통으로 지지되는가? 만약 두 번째 단계에서 어느 정도 유전적 분화가 있었다 할지라도 집단들 사이에 오랜 시간 동안 유전적 교류가 없어서 각 집단이 여러 유전자 상에서 상호단계통성(reciprocal monophyly)을 보유한다면, 이는 종 분화의 역사를 갖는 잠재적 은밀종으로 진단할 수 있다. 여기서 이용되는 계통수는 다수의 유전자들을 이용해 추론한다. 소수의 유전자를 이용한 계통수는 중복, 재조합 등의 원인으로 종 분화의 역사를 반영하기 부적절할 수 있지만, 다수의 유전자에서 동일하게 그런 일들이 발생할 가능성은 낮기 때문이다. 또한, 분자계통수는 그 자체로 생물학적 종개념을 지지하는지 여부를 판정하는데 도움을 준다. 만약 서로 교잡이 가능한 종이었다면, 집단 유전학적 분석을 통해 야생에서 벌어지는 교잡과 불완전한 생식적 격리를 쉽게 검증할 수 있다. 이런 관점을 바탕으로 현재 분자계통학자들은 종간 경계설정(species delimitation)을 위한 방법론을 개발하여 다양한 분류군에 적용하고 있다 [26, 27, 28]. 위와 같은 분자계통학적 연구를 통해 최근 국내에서 발견되는 은밀종의 대표적인 사례는 주로 척추동물의 어류와 양서류에서 찾아볼 수 있다. 우선, 한국의 남부 지역에는 다양한 종의 Hynobius 속의 도롱뇽이 분포하고 있는데 [29, 30], 현재 이 도롱뇽의 무리는 적어도 4종 이상이 존재하는 것으로 발표되었고, 신종 명명이 된 종은 그 중 고리도롱뇽(H. yangi), 제주도롱뇽(H.
quelpaertensis), 꼬마도롱뇽(H. unisacculus), 도롱뇽(H. leechii)이 있다. 이들 종은 매우 미세한 형태적 차이를 갖고 있어서 구분이 쉽지 않은 대표적인 은밀종 무리로 볼 수 있고, 육안 관찰로 이 종을 옳게 구분하는 것은 거의 불가능에 가깝다.
그림 3. 도롱뇽(Hynobius leechii, 좌)과 꼬마도롱뇽(H. unisacculus, 우) [출처: 전형배].
필자와 동료들이 2014년 발표한 낙동납자루(Tanakia latmiarginata)는 담수어류에 있어서 은밀종의 대표적인 사례라 볼 수 있다 [31]. 이 종은 칼납자루(T. koreensis)와 형태적으로 흡사하여 같은 종으로 여겨져 왔으나, 미세한 형태적 차이, 근연종과 독립적인 진화적 역사를 갖는 단계통으로 지지된 다는 점, 칼납자루와 인공 수정 시 발생이 진행되지 않는다는 점을 근거로 신종으로 발표되었다 [31]. 이름처럼 낙동납자루는 낙동강(그리고 수영강)에서 분포하고 형태적으로 유사한 칼납자루는 그 밖의 금강, 섬진강, 탐진강, 만경강 등에 분포하여 분포가 중복되지 않는 것으로 알고 있었으나, 신종 발표 이후 낙동강의 지류인 밀양강에서도 칼납자루의 집단이 발견되었다 [32]. 두 종간의 야생에서의 생식적 격리를 검증할 수 있는 기회였기에, 두 집단의
생명 다양성 보전에 활용되는 보전유전학 연구동향 전형배 Page 9 / 38 유전적 교류가 있었는지를 microsatellites 마커로 검증해보았는데, 예상대로 야생에서도 두 종간 교잡의 증거는 발견되지 않았다 [33]. 결과적으로 칼납자루와 낙동납자루는 분포가 중복되는 지역에서 자연적인 교잡 개체가 출현하지 않고, 인공수정을 통해서도 교잡 개체의 발생이 진행되지 않기 때문에 생식적 격리가 완료된 별개의 종으로 확인되었다. 다행히 낙동납자루는 아직 야생에 많은 집단이 유지되고 있어서 멸종 위기인 수준은 아닌 것으로 보인다. 하지만, 낙동강에서만 서식하는 낙동납자루가 발견되었기 때문에 낙동강의 집단이 별도의 보전 전략이 필요한 보전단위라는 사실을 알게 되었다. 그림 4. 대표적인 은밀종 그룹인 Tanakia 속의 담수어. 좌측상단부터 시계방향으로 칼납자루(T. koreensis), 낙동납자루(T. latimarginata), 임실납자루(T. somjinensis), 묵납자루(T. signifier).[출처: 전형배] 은밀종을 발굴하는 것은 단순히 보전해야 할 생명 다양성을 파악하는 것 이상의 의미가 있다. 우선, 은밀종은 비교적 최근에 분화되었기 때문에 종 분화 메커니즘을 연구할 수 있는 좋은 모델이다 [23, 25]. 멸종이 생명 다양성을 위협하는 사건이라면 반대로 종 분화는 생명 다양성을 끌어올려 주는 원동력이기 때문에, 종 분화의 메커니즘을 이해하는 것은 생명 다양성을 유지하고 북돋아 줄 수 있는 원리를 이해할 수 있다는 점에서 중요하다. 생명 다양성은 과거에 있었던 종 분화가 누적된 결과물이지만, 너무 먼 과거의 사건을 추론한다는 것은 쉬운 일이 아니다. 은밀종의 대부분은 최근까지 공통 조상(common ancestor)을 공유했던 자매 종(sister species)관계로서, 상대적으로 가까운 과거에 있었던 종 분화 메커니즘을 재구성할 수 있게 한다. 이러한 점은 보다 해상력이 높은 역사를 추론하는데 도움을 줄 수 있다.
또한 은밀종의 발굴은 생태계의 기능적 특성에 대한 보다 완전한 이해를 가능하게 한다. 만약 생태계 내에서 각기 고유한 생태적 지위를 점유하는 2종의 은밀종이 있는데, 그 2종을 1종으로 취급한다면, 각 종의 고유한 생태적 특성을 과소평가하게 될 것이다. 예를 들어, 서로 다른 특정한
생명 다양성 보전에 활용되는 보전유전학 연구동향 전형배 Page 10 / 38 숙주에만 기생하는 숙주 특이성을 갖는 기생성 벌 3종이 한 종으로 다루어진다면, 그 종은 각각의 특수한 숙주를 이용하는 specialist들이 아니라 여러 숙주를 이용하는 generalist로 오해하게 될 것이다. 이런 생물의 기능적 특성에 대한 오해는 잘못된 보전 전략을 낳을 수 있다. 이처럼 최근의 분자계통학 연구는 거의 전 생물 분류군으로 확대되며 그동안 밝혀지지 않았던 은밀 다양성을 발굴해 나가고 있다. 하지만, 여전히 아직 발견되지 않은 다수의 은밀종이 존재하는 것 역시 사실이다. 이들 은밀다양성을 보다 효과적으로 발굴하는 것은 보전유전학이 앞으로 풀어나가야 할 숙제 중 하나일 것이다.
2.1.3. 보전 단위(Conservation units)와 보전의 우선순위(Conservation priority) 선정
인간에 의해 야기된 환경변화에도 불구하고 어떤 생물종은 집단의 크기가 감소하지만 어떤 종은 안정적인 생존을 이어나간다. 만약 보전이 필요하지 않은 생물에 대해 보전 전략을 수립하고 실행한다면, 실제 보전이 시급한 종을 보전하지 못하게 될 수 있다. 따라서 보전이 필요한 단위를 규정하고, 보전의 우선순위를 지정할 필요가 있다. 이 보전 단위(conservation units, CUs) 설정과 우선순위(conservation priorities, CPs) 평가는 점차 그 중요성이 증가되어 가고 있음에도 불구하고 여전히 그 보전의 대상은 종의 범주로 규정되는 경우가 대부분이다. 종을 보전의 단위로 설정하는 것은 한계를 갖고 있다. 아직 다른 종으로 분화되지 않았지만, 은밀종과 마찬가지로 같은 종 내에서 유전적으로 독립적으로 분화된 집단들이 존재하는 경우 그 집단들은 보전의 수혜를 입지 못하게 된다. 이 집단들이 중요한 이유는 이들이 결국 생명 다양성을 생성시킬 수 있는 종들로 분화할 여지가 있는 집단들이기 때문이다. 만약 이 지역 집단들이 절멸해버린다면, 앞으로 잠재적으로 분화될 종들의 가능성을 모두 소거해버리는 상황을 초래할 수 있다. 생물의 멸종을 막는 것 만큼이나 중요한 것은 새로운 종이 분화할 잠재성을 보장하는 것이다. 따라서, 종 다양성을 생성시킬 수 있는 잠재력을 유지하기 위해, 한 종 안에 존재하는 집단 수준에서 보전 단위가 설정되어야 할 필요성이 점차 증가되고 있다. 종 하위에 설정될 수 있는 보전의 단위에 대해서는 그동안 몇 가지 기준이 제시되어 왔지만, 현재 가장 널리 받아들여지는 보전의 단위로는 ESUs (Evolutionarily significant units)가 있다 [34, 35]. ESUs는 연구자마다 다양하게 규정하고 있지만 [35], 대체적으로 개념적 정의는 공통적으로 서로 독립적인 진화적 역사를 반영하는 미토콘드리아, 엽록체와 같은 세포소기관(organelle)에 존재하는 유전체와 핵 유전체(e.g. microsatellites) 모두에서 단계통으로 지지되는 각 지역 집단들을 보전 단위로 설정한다는 공통점을 갖고 있다 [35]. 이는 각 집단들이 과거부터 현재에 이르기까지 서로 독립적인 진화적 역사를 갖고 있을 가능성을 시사한다 [34, 35, 36]. 소기관 유전체, 이를테면 미토콘드리아 유전체에 존재하는 변이들은 대부분 선택적으로 중립 (neutral)이며 [37], 일반적으로 모계유전을 따르는 하플로타입 상태로 존재하기 때문에 역사적으로 누적되어온 변이들을 갖고 있다는 특징이 있다. 따라서 이들 분자 마커는 상대적으로 더 먼 과거의 진화적 격리를 설명하기에 적절하다.
생명 다양성 보전에 활용되는 보전유전학 연구동향 전형배 Page 11 / 38 ESU가 과거와 현재까지의 연속적인 분화를 고려하는 개념이라면 관리 단위(management units, MUs)는 그보다는 최근에 격리된 집단들을 규정하는 개념이다 [35]. 다시 말해, microsatel-lites와 같은 최근의 분화를 반영하는 좌위의 대립 유전자 빈도 상에서 차이는 관측되지만, 미토콘드리아 좌위에서는 뚜렷한 분화가 없었을 때 우리는 각각의 집단을 MUs로 제안하게 된다 [35].
MU와 ESU를 평가하기 위해 사용되는 분자 마커로는 microsatellites가 있다. microsatellite은 게놈에서 GAGAGAGAGAGA와 같이 특정한 염기가 반복되는 서열을 말한다. 유전학자들은 이 반복 서열이 반복되는 구간이 게놈 전체적으로 산재되어 있다는 사실을 발견한다 [38, 39, 40]. 이 반복 서열은 일반적으로 DNA 복제 과정에서의 미끄러짐(replication slippage)에 의해 주로 일어나는데 [39], 이 변이는 개체마다 반복 횟수의 차이가 나타날 정도로 해상력이 뛰어났기 때문에 개체 수준의 유전형, 이를테면 친자 분석과 가계도를 작성하는데도 활용된다 [40]. 여러 좌위의 Microsat-ellites 를 활용하면, 집단들 사이의 격리와 유전적 교류를 평가할 수 있다. 이를 위해서는 중립 변이로부터 확보된 유전형 데이터를 이용해 STRUCTURE, PCA, assignment test, Mantel test와 같은 분석을 수행하는 것이 일반적이다. STRUCTURE는 집단간 유전적 구조를 각각의 클러스터로
시각화하여 보여주며 [41], PCA는 집단 간 구조의 가장 설명력 있는 벡터들로 시각화해준다. Assign-ment test는 집단의 평균적인 유전형을 추론한 뒤, 각각의 개체가 본래의 집단에 포함되는지 여부를 검증한다 [42]. Mantel test는 집단들 사이의 지리적 거리가 멀어질수록 유전적 분화도가
증가하는지에 대한 Isolation by distance 가설을 검정하는데 이용된다 [43, 44]. 또한, 근연종에서도 microsatellites 좌위의 데이터를 확보할 수 있다면, New Hybrids와 같은 분석 도구를 이용해 잡종을 분석하는 것도 가능하다 [45]. 이 분석은 잡종의 F1, F2, back cross를 추정치로 보여준다 [45].
한편 최근의 유전체 분석 기술의 발전에 힘입어 집단유전체(population genomics) 분석이 가능해지면서, RADseq (restriction site associated DNA sequencing), GBS (genotype by sequencing)와 같은 RRS (reduced representation sequencing)와 whole-genome sequencing (WGS) 활용해 보다 더 많은 SNPs 좌위를 확보하여 집단 사이의 격리와 유전적 교류를 평가하고 있다 [46, 47, 48]. 이들 기술은 기존 방법보다 더욱 많은 좌위들을 분석할 수 있다는 점이 가장 큰 장점이다. 또한, 집단 수준의 분석을 할 때 적은 개체만으로도 집단 수준의 분화를 설명하는데 용이하다는 장점이 있다. Microsatellites의 경우 집단 간 유전적 구조를 규명하기 위해 집단 별로 충분한 수의 개체를 확보해야 하지만, 집단 유전체 분석은 더욱 많은 수천 혹은 수만 개의 좌위의 SNPs을 분석하기 때문에 적은 개체만으로도 분석이 가능하다. 실제로 소수 개체의 RRS를 분석한 결과와 다수 개체의 microsatellites (13좌위)를 비교한 연구에 따르면, RRS 분석에서는 microsatellites에서 분석에 이용한 개체수의 단 17% 만을 사용했음에도 유전적 구조와 계통 지리적 패턴에 있어서 높은 해상력을 갖는 것으로 나타난 사례도 있다 [49]. 이는 멸종 위기종처럼 개체수 확보가 쉽지 않은 경우에 큰 장점이 될 수 있다 [50]. 한편, 집단 유전체 분석 기술이 갖는 또 다른 장점 중 하나는 바로 자연선택의 영향을 받았을 것으로 여겨지는 후보 유전자(candidate genes)를 이용한 유전적 구조를 설명하게 해준다는 점이다 [51, 52, 53]. 앞서 설명했듯이, 집단유전체에서는 전 유전체에서 고르게 변이들을 추출하기 때문에 특정한 집단의 자연 선택에 대한 흔적을 보여주는 후보 유전자를 발굴할 가능성이 월등히 높다 [53, 54]. 최근에는 후보 유전자를 이용한 지역 적응을 제시하는 연구가 활발히 이루어지고
생명 다양성 보전에 활용되는 보전유전학 연구동향 전형배 Page 12 / 38 있으며 [55], 이 지역 적응을 활용해 보전 단위를 설정하기 위한 사례도 점차 늘어나고 있다. 하지만, 여전히 microsatellites보다는 큰 비용을 지출해야 한다는 단점이 있다. 관리가 필요한 대부분의 야생동식물에 이 방법을 적용하기 위해서는 비용적인 한계를 극복해야 할 것이다. 이제 ESU와 MU의 규정은 여러 멸종 위기종 분류군에서 밝혀지기 시작하고 있고, 한국에서도 이것을 위한 사전 연구가 이루어지고 있다. 충분한 연구가 뒷받침된다면, 머지않은 장래에 보전유전학의 연구성과를 기반으로 한 근거 기반의 보전 전략이 수립될 것으로 기대된다. 보전이 필요한 생물을 규정하는 것 못지않게 중요한 것은 그 종이 살고 있는 서식지를 보전하는 것이다. 생명 다양성의 유지를 위해서는 무엇보다 안정적인 서식지 유지가 관건이다. 멸종을 야기하는 가장 주된 원인은 서식지의 손실이기 때문이다. 하지만, 현실적으로 모든 서식지를 유지 관리한다는 것은 불가능에 가깝기 때문에 일부 중요도가 높은 지역을 보전 지구로 묶어 관리하는 것은 생명 다양성을 유지하기 위한 효과적인 보전 전략으로 활용되어 왔다. 하지만, 그동안 보전 지역의 평가는 객관적인 데이터 기반의 평가가 아닌 전문가 집단의 주관과 직관에 의존하는 한계가 있었다. 최근에는 계통학적 거리(phylogenetic distance)를 이용하여 보전의 우선순위를 평가하는 방법론이 보급되고 있다 [56, 57, 58]. 이 방법은 어떤 지역을 구성하는 종과 개체들이 대체로 먼 계통학적 거리를 갖는다면, 보전의 중요도가 높은 지역으로 평가한다. 먼 계통학적 거리를 갖는다는 것은 그 지역에 존재하는 종들 대부분이 상당한 진화적 역사를 공유할 가능성을 시사하며, 반대로 가까운 계통학적 거리를 갖는 종과 개체들로 구성된 지역은 진화적으로 최근 형성된 집단과 군집일 가능성을 보여준다. 오랜 진화적 역사를 갖는 종과 집단들이 존재하는 지역은 오랜 기간 안정적인 서식지로서 기능해왔을 가능성을 보여주며, 그렇게 오랜 기간 동안 분화된 흔적을 갖는다는 것은 장래의 종 분화의 가능성을 통해 새로운 생명 다양성을 생성할 높은 가능성을 보여주는 것이므로 보전의 가치가 높다. 흥미로운 점은, 보전 가치가 높게 평가된 지역들이 실제 보호 지구로 지정된 곳과 상당히 불일치한다는 사실이다. 이러한 사실은 보전지역 및 보전의 우선순위에 해당되는 집단의 평가가 실제 정책에 반영되어야 할 필요성을 보여준다. 2.2. 유전적 다양성 평가 2.2.1. 중립 변이 중립 변이는 말 그대로 자연선택의 유리함 불리함 어느 한쪽에 편향되지 않은 중립적 성향을 갖는 변이들을 말하며, 유전체에 존재하는 변이 대부분은 (선택적으로) 중립적인 변이들로 알려져 있다. 생존에 영향을 주지 않는 중립 변이들을 집단의 생존을 평가하는데 이용하는 것은 다소 모순적으로 들릴 수 있다. 하지만, 유전적 다양성 평가에 있어서 중립 변이는 진화의 원동력 중 하나인 유전적 부동을 추적하기 위한 분자 마커로 활용 가치가 높다. 비록 중립 변이만으로는 어떤 후보 유전자의 대립 유전자 빈도가 편향되었는지 알 수 없지만, 유전적 부동과 관련된 원인들을 규명할 수 있다. 한편, 유전적 부동은 작은 유효 집단 크기를 갖는 집단에서 단기간 내에 대립 유전자의 빈도를 편향시키는 위력을 발휘할 수 있기 때문에 이를 분석하는 것은 특히 규모가 작은 집단의 보전을 위해 중요한 가치를 지닌다 [59].
생명 다양성 보전에 활용되는 보전유전학 연구동향 전형배 Page 13 / 38 만약 어떤 집단이 산불, 자연재해, 인위적 교란 등의 원인으로 심각한 병목현상을 겪었다면, 집단에 존재하는 개체들이 갖고 있는 중립 좌위들 사이의 연관 불평형(linkage disequilibrium, LD)은 전보다 증가될 것이다. 한편, 적응 변이가 존재하는 좌위는 유전체 내에 존재하는 빈도가 중립 변이 좌위에 비해 적은 반면, 중립 변이는 종내 집단과 종 사이에 변이가 풍부하게 유지되는 특성을 갖고 있기 때문에 유전적 부동을 평가하는 것이 적응 변이보다 유리하다는 이점을 갖고 있다.
2.2.1.1. 유효 집단 크기(Effective population size)
어떤 집단에서 다음 세대의 재생산에 기여하는 개체 수를 유효 집단 크기(effective popula-tion sizes, Ne)라 한다 [60, 61]. 유효 집단 크기는 집단이 현재 처한 상황을 직접적으로 평가할 수 있는 유용한 지표로 널리 활용되고 있다. 이 유효 집단 크기의 측정은 중립 변이를 이용해 측정된다. 중립 변이들은 진화의 원동력 중 하나인 유전적 부동에 깊게 관련되어 있으므로, 유전적 부동을 통해 축적된 중립적인 좌위들 간의 연관 불평형(linkage disequilibrium, LD)의 정도로 유효 집단 크기를 평가할 수 있다(LDNe method, [60]). 이 방법은 작은 유효 집단 크기를 갖는 집단은 보다 강한 유전적 부동의 압력을 받게 되므로, 더 많은 좌위들이 연관 불평형 상태에 놓일 것이라는 가정에 근거한다. 만약 어떤 집단에서 다수의 좌위가 연관 불평형 상태이고 다른 집단은 그보다 적은 일부의 좌위만이 연관 불평형 상태라 한다면, 전자는 유효 집단 크기가 작게 평가되고 후자는 유효 집단 크기가 크게 평가된다. 물론 집단 내 재조합(recombination)이 발생하는 정도는 비슷해야 이 평가는 유효하다.
유효 집단 크기(Ne)와 실제 집단의 성체 집단 크기(adult census population size, Nc) 사이의 비율(Ne/ Nc)은 집단이 처한 상황을 평가하는데 활용된다 [62, 63]. 만약 집단을 구성하는 성체 집단 크기보다 유효 집단 크기가 현저히 낮다면, 그렇지 않은 경우보다 유전적 다양성을 더욱 빠르게 잃게 될 수 있다 [64]. 성체 집단 크기가 포식, 경쟁, 기생, 교란 등과 관련된 생존력에 의해 결정되는 값인데, 다음 세대의 유전적 다양성에 기여할 개체수가 그보다 현저히 낮다는 것은 쉽게 말해, 집단의 개체수는 많지만 유전적 잠재력은 낮은 “속 빈 강정”이란 의미이다. 따라서, Ne/ Nc를 이용하면 간편하게 집단이 처한 상황을 진단 할 수 있다. 한편, 집단은 여러 세대의 개체들로 구성되어 있기 때문에, 이를 반영한 유효 집단 크기는 시간에 따른 효과를 반영하지 않는다. 따라서, 시간의 변화에 따른 유효 집단 크기의 추정을 위해서는 단일 세대의 유효 집단 크기를 측정하는 것이 효과적이다. 이를 코호트 집단 크기(effective number of breeders, Nb)라 한다 [65, 66]. 성체 집단 크기에 대한 코호트 집단 크기의 비율(Nb/Nc)은 특정 코호트의 보전과 진화적 분석을 위한 기초적인 지수로 활용된다 [67]. 성체 집단 크기가 포식, 경쟁, 기생, 교란 등과 관련된 생존력과 직접적으로 관련되었기 때문에 이보다 낮은 코호트 집단 크기는 일부 개체가 다음 세대를 재생산하는데 기여한다는 다음 세대에 근친교배, 불균형한 성비, 성 선택, 번식 성공도의 편향 등의 영향을 받기 때문에 이 지수의 변동은 위와 같은 변수들의 효과를 검증하기 위한 좋은 출발점이 된다 [67]. 다음 세대의 재생산은 매년 일정하지 않기 때문에 코호트 집단 크기는 해마다 변동이 있을 수 있다. 하지만, 야생 집단의 코호트 집단 크기가 성체 집단 크기보다 월등히 떨어지는 경향이 매 세대를 거치면서 점점 더 심해진다면, 그 원인을 찾기 위해 위에 열거한 후보 원인들(e.g. 근친교배 여부, 성비의 편향 여부, 번식 성공도의 편향 여부, 번식지의 교란 등)을 검증해볼 필요가 있다. 두 수치 사이의 지속적인 격차는 곧 집단이 감소된다는 분명한 신호이기 때문이다.
생명 다양성 보전에 활용되는 보전유전학 연구동향 전형배 Page 14 / 38 유효 집단 크기는 현재의 진단뿐만 아니라 보전 전략의 유효성을 평가할 수 있는 지표로써 활용될 수 있다. 기존의 보전 전략 평가 방법은 대부분은 생물상을 주기적으로 모니터링하고 종의 숫자와 개체수가 늘어나면 효과적인 보전 전략이었다고 평가해왔다. 하지만, 이러한 재래식 방법은 몇 가지 함정이 도사리고 있다. 만약 종의 숫자와 개체수가 증가한 것을 긍정적으로 평가한다면, 다른 지역에서 이주, 이식에 의해 종과 개체수가 증가한 것을 긍정적으로 평가할 수 있고, 인위적인 결과 값의 왜곡을 막을 수 없다. 유효 집단 크기와 성체 집단 크기는 현재의 집단 크기와 장래의 집단 크기의 잠재력을 동시에 평가할 수 있으므로 보다 현실적이고 왜곡될 가능성이 낮은 지표이다. 특히 유효 집단 크기는 단순히 집단의 개체수가 증가된다고 즉각적으로 변동이 일어나지 않기 때문에 보다 안정적인 지표로서 보전 전략의 사후 평가에 활용될 수 있다. 2.2.1.2. 집단 크기의 역사적인 변동(Demographic history) 중립 변이들은 집단이 분화할 당시의 변이들을 유전체 내에 보존하고 있기 때문에 최근의 유전적 부동에 영향을 주는 사건들 뿐만 아니라 역사적인 사건들을 설명하는데 활용된다. 최근의 집단 유전체 분석 기술의 발전으로 보다 많은 SNPs들을 분석에 활용할 수 있게 되면서, 과거로부터 보존적으로 유전체에 남아있던 변이들을 활용해 과거의 집단이 겪었을 집단 수준의 사건들
이를테면 병목현상(bottleneck), 집단의 급격한 팽창(population expansion), 집단 간 유전적 흐름(gene flow) 등을 규명할 수 있게 되었다 [68, 69].
집단 크기의 역사적인 변동(demographic history)은 진화적 모델을 주어진 유전적 데이터를 바탕으로 시뮬레이션을 수행해 가장 유의미한 모델을 선정한다. 집단의 진화 모델은 집단의 감소와 팽창을 나타내는 유전적 병목 모델(bottleneck), 집단 사이의 유전적 흐름을 가정하는 모델(gene flow), 집단 간 분화를 가정하는 모델(divergence) 등이 있다 [69, 70]. 각 모델들은 합류
이론(coalescent theory)에 근거한 방법, 근사 베이즈 계산(approximate Bayesian computation, ABC)과 같은 베이즈 추론(Bayesian inference)을 이용한 방법, 끝으로 최근 각광받는 확산이론에 근거한 SFS (site frequency spectrum)을 이용한 방법을 통해 평가된다 [69, 71]. 특히, SFS를 활용해 집단의 역사를 추론하는 것은 비단 중립 변이에 의한 역사뿐만 아니라, 자연 선택의 압력을 받는 변이를 규명하는 데에도 활용된다 [71]. 2.2.1.3. 집단의 유전적 구조(Population structure) 위의 모델 기반 방법들 이외에 집단의 진화적 역사와 계통을 규명하기 위해 비-모델 기반의 방법을 활용해 집단의 유전적 구조를 규명할 수 있다. 대표적인 분석 도구로는 STRUCTURE [41], ADMIXTURE, sNMF [73] 등의 분석 방법이 있다. 모델 기반 방법과 달리 이들 방법은 집단 크기의 증감을 설명하는 데는 한계가 있다. 그럼에도 집단의 격리와 이주의 흔적을 개체 수준에서 세밀하게 검증할 수 있다. 만약 집단을 구성하는 개체들 중 다른 집단의 유전적 특성을 갖는 개체가 존재할 경우 그 개체는 최근의 유입의 역사를 반영할 것이며, 만약 집단의 유전체에 고르게 다른 집단으로부터 유전적 교류의 흔적이 관측될 경우, 이는 좀 더 과거의 유전적 교류의 역사를 보여줄 것이다. 반대로 유전적으로 격리된 역사 역시 이 방법으로 확인할 수 있다. 이 방법을 인간의 집단에 응용하면, 어느 지역의 집단이 어떤 유래를 갖고 얼마나 유전적으로 교류해 왔는지에 대한 정보를 얻을 수 있다. 실제로 영국에서 인간 집단을 대상으로 이와 같은 연구가 이루어지기도 했다.
생명 다양성 보전에 활용되는 보전유전학 연구동향 전형배 Page 15 / 38 그림 5. microsatellites 좌위로 분석된 한반도 남부 Tanakia 종의 유전적 구조 [33]. Tanakia koreensis의 경우 2개의 유전적 구조가 확인된다. 2.2.1.4. 과거에 분화된 분류군 사이의 유전적 흐름 한편, 과거로부터 전해져 내려온 유전적 격리의 증거는 계통수를 이용한 분석을 통해서도 손쉽게 검증할 수 있다. 그런데 기존의 계통 분석은 각 집단(계통) 사이에 존재했던 과거의 유전적 흐름을 규명하기에는 취약하므로, D 통계량(D statistics 혹은 ABBA-BABA statistics) 분석을 통해 과거의 조상 단계에서 있었을 집단 혹은 분화된 종 사이의 유전적 교류를 평가할 수 있다 [74, 75]. D 통계는 조상형(ancestral)의 유전형으로부터 파생된(derived) 유전형이 임의로 존재한다는 가정에서 출발한다. 이 파생형과 조상형은 선택적으로 중립(neutral)이기 때문에 각 집단들에 균등하게 분포할 것으로 예측된다. 만약 어떤 종이 과거에 다른 종으로부터 조상형 혹은 파생형의 대립 유전자를 받아들인, 다시 말해 유전적 흐름(gene flow)이 존재했었다면, 그 분포에 편향이 발생될 것이기 때문에 이 편향을 파악해 분화된 그룹 간 유전자 이입의 정도를 파악할 수 있게 된다. 2.2.1.5. 가계분석 야생 상태에서 매우 제한적인 크기를 갖거나, 인위적인 공간에서 사육되는 집단들은 가계 분석을 통해 집단의 상황을 모니터링하고 효과적인 관리를 위해 활용될 수 있다. 여기에도 마찬가지로 중립 좌위들이 활용된다. 만약 적응 좌위를 이용한다면, 정화 선택(purifying selection)을 유발하는 강력한 치사 유발 대립 유전자와 같은 문제로 인해 가계 분석의 효율에 문제가 생길 수 있다. 인간을 비롯해 많은 생물들의 가계 분석을 위해 오랫동안 애용되어온 분자 마커는 microsatel-lites이다 [75, 76]. 연구하고자 하는 종에서 변이가 충분한 마커만 개발되어 있다면, 적은 비용으로도 많은 개체들의 가계 분석이 가능한 장점이 있다 [69, 75]. 지난 20여 년 동안 분자생태학자들은 거의 모든 식물과 동물의 분류군에 적용할 수 있는 microsatellites 마커를 개발해왔기 때문에 특수한 분류군을 제외하면 가계 분석은 microsatellites만으로도 충분히 가능하다. 게다가 가계 분석을 위해서는 다른 분석들과 달리 그리 많은 좌위가 필요치 않다는 점도 여전히 microsatellites의 유용성에 한몫한다.
생명 다양성 보전에 활용되는 보전유전학 연구동향 전형배 Page 16 / 38 하지만, 최근 발전하고 있는 NGS 기술은 가계도 분석에서도 그 위력을 발휘하고 있고, 특히 유전체 영역에 광범위하게 존재하는 SNPs들 중에서 적절한 변이를 갖는 후보 SNPs 마커를 개발한다면, microsatellites 방법보다 낮은 가격으로 가계 분석이 가능하게 되었다 [77]. 그림 6. 가계 분석을 위한 방법과 그 비용 [77]. 2.2.2. 적응 변이 비록 대부분의 변이들이 중립적이지만, 일부 변이들은 적응과 관련되어 있다. 적응 변이는 말 그대로 집단이 해당 환경에 적응하고 생존하는데 직접적으로 관련되어 있는 변이이기 때문에 이 변이들에 유해한 변이가 누적된다면, 이는 집단의 존립을 위협할 수 있다. 이런 이유로 적응 변이 연구의 중요성은 인간에 의한 급격한 환경 변화에 따른 제6의 대멸종의 시대에 접어든 현세에 들어 더욱 커지고 있다 [53, 54]. 중립 변이 만을 가지고는 어떤 특정 집단이 실제로 어떤 위협에 처해있고 어떻게 대응을 해야 할지에 대한 해답을 제시하는 게 매우 제한적이기 때문이다. 2.2.2.1. 후보 유전자(Candidate genes) 적응과 관련된 후보 유전자들은 모델 생물을 기반으로 한 연구들을 통해 발굴되어 왔다. 유전체 분석 기술의 괄목할만한 발전은 비-모델생물로 후보 유전자 발굴 연구를 확장시켜 나가고 있다. 이렇게 발굴된 적응 유전자는 환경과 지역 별로 달리 나타날 수 있다. 이를 테면 여우에게 흰털/ 검은털을 만드는 좌위가 단일 좌위라면, 흰털이 은신에 이점을 제공할 가능성이 높기 때문에, 눈 덮인 지역에서는 흰털과 관련된 대립 유전자의 빈도가 높거나 고정되어 있을 것이다. 척추동물에서 기존의 유전학적 방법론으로 연구되어온 전통적인 적응 유전자는 MHC 유전자가 있다 [78]. 외부에서 유래된 항원 펩티드 조각으로부터 비자기를 인지하는 적응 면역 과정에 관여하는 이 MHC는 크게 class I, II 그리고 III 가 있는 것으로 알려져 있다 [79]. class II는 전문적인 항원 제시 세포(antigen-presenting cells, APCs)의 세포 표면에 존재하며, class I은 모든
생명 다양성 보전에 활용되는 보전유전학 연구동향 전형배 Page 17 / 38 세포 표면에 존재한다. 기능적으로 이 둘은 차이를 갖는데, class II는 박테리아와 같은 외부 항원을 포식한 항원 제시 세포가 분해된 항원 유래 펩타이드 조각을 CD4+ helper T 세포에 제시하는 경로를 갖지만, class I은 바이러스에 의해 감염된 세포에서 감염원인 항원의 정보를 CD8+ cytotoxic T 세포에 제시하는 차이가 있다 [79]. 바이러스에 의한 세포의 감염은 특정 세포에 국한하지 않고 무작위적으로 발생되기 때문에 Class I은 모든 세포에 존재하는 것으로 추정된다. 구조적으로도 이 두 class는 차이가 있는데, class II는 각 두 개의 도메인으로 구성된 알파 사슬과 베타 사슬의 heter-odimer 형태로 이루어져 있는 반면 class I은 세 개의 알파 사슬과 베타 마이크로글로불린으로 구성되어 있다. 항원 유래 펩티드가 결합하는 부위는 class I의 경우 알파1과 알파2 도메인이지만, class II의 경우 알파1과 베타1 도메인에 해당한다 [80]. 그림 7. MHC class II beta의 단백질 3차구조 [33]. 자주색은 항원에서 유래한 펩티드가 결합하는 부위(PBR)를 가리킨다. MHC 유전자의 특징은 유전체 내에서 가장 많은 변이를 갖는 다형적인(polymorphic) 영역이라는 점이다. 인간의 HLA-B 대립 유전자의 가짓수는 1,000개가 넘는다 [81]. 참고로 필자가 동료와 함께 한 종의 물고기에서 발굴한 MHC class II 대립 유전자의 숫자는 140개였다 [82]. 척추동물에서 어떤 유전자도 이 정도의 다형성을 갖고 있지 않다. 그중에서도 특히 항원과 결합하는 잔기인 항원 결합 부위(peptide binding region, PBR)에 변이가 집중되는 경향을 보여준다 [82].
이러한 높은 다형성은 유전자 중복(duplication), 재조합(recombination) 등을 통해 나타나며 [82, 84], 한번 만들어진 변이들은 병원체와의 공진화(pathogen-mediated selection [85]), 배우자 선택(mate choice [86, 87]), 유전자 이입(introgression [88]), 음성 빈도 의존적 선택(negative fre-quency dependent selection [89]) 등의 메커니즘을 통해 집단 내에 사라지지 않고 유지된다. 사라지지 않은 적응 변이들은 이따금씩 진화적으로 먼 거리의 분류군 사이에도 보존되는 경향을 보여준다. 이를 “trans-species polymorphism [90]”이라고 한다. MHC 유전자는 보전유전학적 가설을 검증하기 위해 사용되어 왔다 [1, 91, 92]. 적응 변이는 집단의 크기 감소와 관련되어 있을 것으로 추정할 수 있다 [91]. 실제로 집단의 크기가 급격히 감소된 종 혹은 집단의 MHC 대립 유전자 다형성은 그렇지 않은 종 혹은 집단에 비해 낮은 경향을 갖고 있었다 [93, 94]. 한편, 이렇게 감소된 MHC 다양성은 질병에 대한 취약성과 뚜렷한 관련성이
생명 다양성 보전에 활용되는 보전유전학 연구동향 전형배 Page 18 / 38 없다는 연구 결과도 있어서 [95], MHC 다양성의 감소가 병원체에 대한 저항성과 관련이 있는지는 더욱 많은 증거가 필요한 상황이다 [95]. 병원체와 관련된 대립 유전자들이 연구되어온 인간, 가축, 실험 동물과 달리, 병원체와 연관된 대립 유전자의 변이가 밝혀지지 않은 야생동물의 경우에 특히 신중함이 요구된다. 아직 야생생물들을 대상으로 MHC 다형성을 고려한 보전생물학적 연구는 연구 방법의 난점으로 인해 많지 않은 상황이지만, 향후 지속적인 연구가 수행되면 생물들이 처한 상황에 대한 더 면밀한 평가와 대응 전략 마련에 도움이 될 것이다. 적응 변이는 자연 선택의 영향을 받기 때문에 환경에 의해 결정된다. MHC 유전자 역시 마찬가지로 이 유전자의 높은 다형성은 종의 생태적 특성에 의해 결정된다. 철새와 텃새의 MHC 다양성을 비교한 연구에 따르면, 이주하며 다양한 병원체를 접하는 철새가 텃새에 비해 더 MHC 대립 유전자 내에서 아미노산의 변화를 초래하는 비동의 치환(nonsynonymous substitutions)이 더 많이 축적되었다고 한다 [96]. 또한, 무리를 이루고 사는 생물들도 MHC의 다형성이 높은 경향이 있다. 조류에서 연구된 바에 따르면, 단독생활을 하는 종보다 무리를 이루고 사는 종이 보다 높은 수준의 비동의 치환을 갖고 있는 경향이 밝혀졌다 [96]. 무리생활을 하는 박쥐 역시 포유류에서 가장 다양한 MHC 다형성을 보유한 것으로 알려져 있다 [97, 98]. 그리고 다양한 병원체를 보유한 것으로 알려져 있다 [97]. 이런 결과들은 다양한 병원체를 접하는 집단이 보다 다양한 대립 유전자가 형성되어온 균형 선택(balancing selection)이 작동해왔음을 의미한다. 따라서, 제한적인 다형성을 갖는 집단은 적은 수의 병원체에 적응해왔을 것이며, 이들 집단에 외부의 병원체의 유입은 위험한 상황을 초래할 가능성이 있으니 보전 전략 수립이 필요하다는 점을 시사한다. 또한, 다양한 병원체 적응된 유전적 적응을 이해하는 것은 동일한 병원체의 위험에 놓인 인간 집단의 보건을 위해서도 중요할 것이다. 한편, 대립 유전자의 다형성은 비용을 초래할 수 있다. 인간 유전체에 존재하는 MHC의 높은 다형성은 주변의 연관된 유전자에 해로운 변이를 축적되도록 하는 비용을 초래한다고 한다 [99]. 유전체의 유해 변이들은 정화 선택(purifying selection, 혹은 negative selec-tion)을 통해 집단 내에서 소실되는 것으로 알려져 있다. 그런데 정화 선택의 압력은 강한 균형 선택이 작용하는 좌위 주변에서는 상쇄된다는 것이다. 그 결과 유해할 수 있는 변이들이 집단에서 사라지지 않고 유지된다는 것이다. 큰가시고기의 경우에서도 개체가 보유하고 있는 대립 유전자 숫자의 빈도 분포는 최대값이 아닌 중앙값에 수렴된다 [100]. 멸종 위기에 처한 생물들을 보전하기 위한 인공증식 프로그램(captive breeding program)은 MHC의 대립 유전자 다양성을 감소시키는 원인으로 지목되었다 [101]. 연어가 보유했던 편향된 MHC 다양성이 집단의 MHC 대립 유전자의 빈도를 왜곡시키는 사례가 증명되었고, 인위적으로 증식된 브룩송어(brook trout)는 성장율에 대한 선택압이 가해지기에 그 반대급부(trade-off)로 MHC 변이에 대한 선택압이 낮아지기 때문에 양식으로 길러진 개체의 방류는 적응 변이의 측면에서 그리 바람직하지 않음이 제시되기도 하였다 [102]. 한편, 최근의 NGS를 이용한 유전체 분석에 힘입어 다양한 생물 분류군에서 후보 유전자를 발굴하는 연구가 활발하게 이루어지고 있다 [103, 104, 105, 108]. 이들 후보 유전자들 중 야생의 집단에서 발견되는 다형적인 후보 유전자들은 집단의 진화적 변화를 추적할 수 있는 분자 마커로 활용될 수 있다 [105]. 북반구 고위도 선을 따라 전 세계적으로 분포하고 진화생물학의 모델 생물로 활용되는 큰가시고기에서 밝혀진 후보 유전자들은 이 작은 물고기가 환경 변화에 어떻게 적응해왔는지 설명하는데 활용되고 있다 [106, 107]. 큰가시고기는 이름처럼 지느러미에 큰 가시를 갖고 있고 뿐만 아니라 몸통에 넓은 뼈로 된 인판을 갖고 있다. 인판과 가시는 포식자로부터 몸을
생명 다양성 보전에 활용되는 보전유전학 연구동향 전형배 Page 19 / 38 보호하는 기능을 하는데, 이 가시와 인판의 발달과 관련된 후보 유전자의 대립 유전자의 빈도는 이들이 살아가는 서식지에 따라 변화한다. 따라서 후보 유전자의 대립 유전자 빈도를 통해 각 집단의 적응도를 평가할 수 있게 된다 [107]. 그림 8. 큰가시고기에서 밝혀진 후보 유전자들 [107]. (A) 멜라닌 색소 침착과 관련된 KITLG, (B) 이빨 숫자의 후보 유전자 BMP6, (C) 인판 발달과 무리 행동에 관련된 EDA, (D) 인판 발달에 관련된 GDF6. 그림 9. 인위적으로 개량(domestication)된 금붕어의 형태에 연관된 후보 유전자들 [108].
생명 다양성 보전에 활용되는 보전유전학 연구동향 전형배 Page 20 / 38 2.2.2.2. 유해 변이의 축적(Accumulation of deleterious mutations)
적응과 관련된 변이들 중에는 생존에 불리함을 주는 유해 변이(deleterious mutations)들도 존재한다. 이런 유해 변이들은 대부분 단백질을 암호화하는 유전자 서열에서 종결 코돈을 만들어 비정상적인 단백질을 번역하기 때문에 유해 변이가 축적된다면, 개체의 적응도에 불리하게 작용할 수 있다 [109]. 유해 변이는 크게 LOF(loss-of-function, [110]), NMD(nonsense-mediated mRNA decay, [111])를 야기하는 변이들을 식별하여 확인할 수 있다. 유해 변이는 일반적으로 최근의 작은 유효 집단 크기를 갖게 된 집단에 더욱 누적되는 경향을 보여준다 [112, 113, 114]. 한편, 오랫동안 고립되어온 집단들은 작은 유효 집단 크기에도 불구하고 유해 변이의 누적된 정도가 비교적 안정적인데, 이는 정화 선택(purifying selection)에 의해 유해 변이를 가진 개체들이 여러 세대에 걸쳐 도태(purging)되었기 때문으로 여겨진다 [115, 116]. 유해 변이는 집단의 운명에 부정적 영향을 직접적으로 미칠 수 있기 때문에 최근 점차 다양한 야생 집단에서 연구되기 시작하고 있다. 유해 변이의 정량적 분석은 PROVEAN [117]과 SnpEff [118]을 통해 가능하며, 전자는 레퍼런스 유전체(reference genome)가 존재하지 않아도 분석이 가능하지만, 후자는 레퍼런스 유전체 데이터가 존재해야 한다. 따라서 후자는 별도의 annotation 과정 없이 기존 유전체에 mapping된 SNPs를 바탕으로 유해 변이 여부를 즉시 평가할 수 있다는 장점이 있다. 2.2.2.3. 지역 적응(Local adaptation) 집단의 이주가 제한적인 경우 대부분의 적응 변이들은 서식지에 특이적으로 선택된다. 따라서 각 지역 집단 별로 고유한 적응 변이들을 갖는데, 이를 ‘지역 적응(local adaptation)’이라 한다 [119, 120]. 각 분류군 별로 고유한 지역 적응을 규명하기 위해서는 유전체의 고른 영역을 분석할 수 있어야 하므로, 집단 유전체 분석이 보급되기 이전에는 대부분 후보 유전자를 중심으로 연구되어 왔다 [119, 121]. NGS 기술이 야생생물 연구에도 보급되기 시작한 최근 10년 이래로 많은 분류군에서 지역 적응의 증거가 밝혀지고 있다. 성공적인 지역 적응 규명을 위한 관건은 다양한 환경에서 유래된 개체들의 최대한 넓은 유전체 영역을 톺아보는 것이다. 유전체 분석 방법의 비용적 난관이 해결되어감에 따라 점차 유전체 전 영역으로 연구가 확대되고 있다. 2.3. 집단의 유전적 다양성이 유지되는 메커니즘 중립 변이와 적응 변이를 포괄한 전체적인 유전적 다양성이 집단 수준에서 유지되는 메커니즘을 아는 것은 집단의 보전을 위한 효과적인 대책을 마련할 수 있다는 점에서 중요하다. 집단의 유전적 다양성은 다음의 메커니즘에 의해 유지될 수 있다. 첫 번째는 비임의적 교배 시스템이다. 유성생식은 자손들의 재조합 과정을 통해 유전적 다양성을 유지, 증가하는데 기여할 수 있다 [122]. 이와 반대로, 특정한 형질에 대한 임의의 배우자 선호성은 자손들의 유전적 다양성을 감소시킬 수 있는데, 만약 모든 여성이 특정 남성만을 배우자로
생명 다양성 보전에 활용되는 보전유전학 연구동향 전형배 Page 21 / 38 선호하고 그와 자손을 만든다고 가정한다면 다음 세대의 자손들의 유전적 다양성의 폭은 제한될 것이다. 이러한 경향이 여러 세대 동안 이어진다면, 그 집단은 몇 세대 만에 유전적 다양성을 극적으로 소실하게 될 것이다. 이처럼 임의의 교배 시스템은 유효 집단 크기의 감소를 초래하고 집단의 유전적 역동성의 제한을 야기함에도 불구하고 많은 종들에게서 이러한 임의의 배우자 선택이 진화되어 왔다. 특히 공동의 번식 장소인 렉(lek)을 갖는 일부 조류에서 임의 교배는 집단의 유전적 다양성을 감소시킬 심각한 위협이 될 수 있음에도 이런 배우자 선호성이 유지되고 있다. 이를 “렉의 역설(lek paradox)”이라 한다 [123]. 이 역설을 해결하기 위한 연구들이 수십 년째 이어지고 있지만, 아직 명쾌한 답은 내려지지 않고 있다 [123, 124]. 그림 10. 메타집단구조. (출처: https://kevintshoemaker.github.io/NRES-470/LECTURE13.html) 또 하나의 유전적 다양성을 유지하는 기작은 바로 ‘집단 간 유전적 교류’이다. 일부 생물들은 인간의 힘을 빌리지 않고도 자연적으로 이따금씩 이주의 가능성이 열려있다. 예를 들어 고립된 섬은 가끔씩 태풍에 의해 새로운 식물의 씨앗이 이주하기도 한다. 또한, 새의 소화되지 않은 씨앗은 배설물을 통해 기존의 다양성이 떨어져 가는 섬의 집단에 새로운 변이를 공급해줄 수 있다. 실제로 여러 생물들에 있어서 이주에 의해 집단의 다양성이 유지되는 사례들이 보고되고 있다. 그중 이주에 의한 유전적 다양성의 유지를 설명하는 가장 극적인 모델은 바로 메타 집단 모델(metapopulation dynamics)이다 [125]. 메타집단이란, 집단들 각각의 유전적 다양성이 떨어진다 할지라도 그 집단들 사이에 유전적 교류를 통해 전체적인 유전적 다양성이 소실되지 않고 유지되는 상태를 말한다. 예를 들어 작은 섬인 울릉도에 자생하는 어떤 식물은 오래 존속하지 못하고 주기적으로 절멸을 반복하지만, 새의 깃털에 붙은 씨앗을 통해 매년 새로운 세대의 공급이 이루어져 집단이 유지된다면 우리는 그것을 메타집단이라고 규정할 수 있다. 개체의 이주는 서로 균등하게 일어날 수도 위의 사례처럼 한쪽 방향으로 일방적으로 일어날 수도 있다. 후자의 경우 개체의 공급을 받은 울릉도 집단은 “sink”, 개체의 공급이 일어난 내륙의 집단을 “source”라고 부를 수 있다.
![그림 3. 도롱뇽( Hynobius leechii, 좌)과 꼬마도롱뇽( H. unisacculus, 우) [출처: 전형배].](https://thumb-ap.123doks.com/thumbv2/123dokinfo/5123259.86540/8.892.119.778.651.866/그림-도롱뇽-hynobius-leechii-꼬마도롱뇽-unisacculus-출처-전형배.webp)
