저작자표시 2.0 대한민국 이용자는 아래의 조건을 따르는 경우에 한하여 자유롭게 l 이 저작물을 복제, 배포, 전송, 전시, 공연 및 방송할 수 있습니다. l 이차적 저작물을 작성할 수 있습니다. l 이 저작물을 영리 목적으로 이용할 수 있습니다. 다음과 같은 조건을 따라야 합니다: l 귀하는, 이 저작물의 재이용이나 배포의 경우, 이 저작물에 적용된 이용허락조건 을 명확하게 나타내어야 합니다. l 저작권자로부터 별도의 허가를 받으면 이러한 조건들은 적용되지 않습니다. 저작권법에 따른 이용자의 권리는 위의 내용에 의하여 영향을 받지 않습니다. 이것은 이용허락규약(Legal Code)을 이해하기 쉽게 요약한 것입니다. Disclaimer 저작자표시. 귀하는 원저작자를 표시하여야 합니다.
Region-Specific Expression of
Brain-Derived Neurotrophic Factor (BDNF) mRNA
after Long Term Treatment with Lithium
Carbonate
by
Mi Young Eu
Major in Neuroscience
Department of Biomedical Sciences
The Graduate School, Ajou University
Region-Specific Expression of
Brain-Derived Neurotrophic Factor (BDNF) mRNA
after Long Term Treatment with Lithium Carbonate
by
Mi Young Eu
A Dissertation Submitted to The Graduate School of Ajou University
in Partial Fulfillment of the Requirements for the Degree of
Master of Neuroscience
Supervised by
Byoung Joo Gwag, Ph.D.
Major in Neuroscience
Department of Biomedical Sciences
The Graduate School, Ajou University
This certifies that the dissertation of
Mi Young Eu is approved.
SUPERVISORY COMMITTEE
Byoung Joo Gwag
Eun Joo Baik
Yong Beom Lee
The Graduate School, Ajou University
December, 22nd, 2008
i -ABSTRACT-
Region-Specific Expression of Brain-Derived Neurotrophic Factor
(BDNF) mRNA after Long Term Treatment with Lithium Carbonate.
There is much evidence that chronic administration of lithium ion protects neurons in vitro, and in various animal models of neurological diseases. The possibility has been raised that the neuroprotective effects of lithium ion may be mediated through regulation of brain-derived neurotrophic factor (BDNF). Recently, lithium ion has been shown to selectively protect spinal motor neurons in animal models of amyotrophic lateral sclerosis (ALS) by preventing the apoptosis component. In this study, investigation was undertaken to determine if lithium ion would regulate expression of BDNF for its anti-apoptosis action. RT-PCR and fluorescence in situ hybridization experiments were performed to determine the lithium-sensitive expression profile in brain and spinal cord. In the brain, chronic treatment with lithium increased mRNA expression of BDNF in the cerebral cortex, the hippocampal formation, the striatum, the diencephalons, and the midbrain, but not in the cerebellum. In particular, BDNF mRNA expression was exclusively increased in neurons. Long-term treatment with lithium increased levels of mRNA in all regions of the spinal cord including the cervical, thoracic, and lumbar regions. Immunohistochemistry and in situ hybridization revealed that BDNF expression was increased selectively in large spinal motor neurons of mice treated with lithium. The present findings suggest that chronic treatment with lithium
ii
increases expression of BDNF in various populations of neurons in the brain, which underlies the pharmacological action of lithium as a mood stabilizer, and also suggests a novel therapeutic potential as a neurotrophic agent.
Dopaminergic neurons in the substantial nigra were shown to undergo degeneration in G93A transgenic mouse models of ALS. To examine if lithium ion would protect the dopaminergic neurons in ALS mice, loss of the dopaminergic neurons was first verified by counting cells immunoreactive to tyrosine hydroxylase (TH) antibody using a stereology method in 16-week old male G93A mice. There was a 28 % decrease in the number of dopaminergic neurons in the ALS mice. The degeneration of the dopaminergic neurons was prevented by daily administration of lithium in the diet for 4 weeks. In the G93A mice, expression of BDNF mRNA was significantly decreased compared to the wild type. In G93A mice treated with lithium ion, expression of BDNF mRNA was comparable to the control. Thus, lithium ion prevents degeneration of the dopaminergic neurons in G93A mice through mechanisms involving upregulation of BDNF.
Key Words : BDNF, ALS, G93A, ROS, Apoptosis
iii
TABLE OF CONTENTS
ABSTRACT ··· i
TABLE OF CONTENTS ··· iii
LIST OF FIGURES ··· v
LIST OF TABLES ··· vi
ABBREVIATION ··· vii
Ⅰ. INTRODUCTION ··· 1
Ⅱ. MATERIALS AND METHODS ··· 3
A. MATERIALS ··· 3
1. Experimental Animal models ··· 3
B. METHODS ··· 3
1. SYBR Green I reverse transcription-polymerase chain reaction ··· 3
2. Stereology ··· 4
3. Immuohistochemistry ··· 4
4. Fluorescence In situ hybridization ··· 5
5. Fluorescent intensity ··· 6
6. Data analysis ··· 6
Ⅲ. RESULTS ··· 7
A. Lithium increases BDNF mRNA expression after chronic treatment in Brain and spinal cord ··· 7
B. Lithium has an effect on protection neurons from degenerative death in ALS animal model ··· 10
iv
Ⅴ. CONCLUSION··· 27 REFERENCES ··· 28 국문요약 ··· 34
v
LIST OF FIGURES
Fig. 1. Li+ increases BDNF mRNA expression in various brain areas in adult rat ∙∙∙∙∙ 8 Fig. 2. Li+ increases expression of BDNF mRNA in the spinal cord of adult rat. ∙∙∙∙∙ 9 Fig. 3. Li+ increases mRNA and protein expression of BDNF in the lumbar spinal
motor neurons of adult rat. ∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙ 12 Fig. 4. Li+ attenuates degeneration of spinal motor neurons in ALS mice. ∙∙∙∙∙∙∙∙∙∙∙∙∙∙
13 Fig. 5. Li+ increases BDNF expression and Akt phosphorylation in the lumber spinal
cord of adult mice. ∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙ 14 Fig. 6 . Li+ prevents apoptotic neuronal cell death. ∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙
17 Fig. 7. Li+ cannot reduce ROS level and cannot prevent ROS-mediated cell death.
∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙ 20 Fig. 8. Li+ increases expression of BDNF mRNA in the substantia nigra in ALS
vi
LIST OF TABLE
Table 1. The mean total number of TH positive cells in the unilateral substantia nigra
of the 16 weeks old male G93A mouse.
∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙∙
15vii
ABBREVIATION
ALS : Amyotrophic lateral sclerosis
BDNF : Brain-Derived Neurotrophic Factor
BPD : bipolar disorderDAB : 3,3’-diaminobenzidine tetrahydrochloride dehydrate Li+ : Lithium ion
PBS : phosphate-buffered saline
PI3K : phosphatidylinositol 3-kinase SN : substantia nigraTH : tyrosine hydroxylase
1
Ⅰ. INTRODUCTION
Evidence has accumulated showing that chronic administration of Li+ protects neurons in
vitro (Kang et al., 2003) and in various animal models of neurological diseases. Chronic treatment with lithium increased BDNF expression in the rat frontal cortex and hippocampus (Fukumoto et al., 2001).
The possibility has been raised that the neuroprotective effects of Li+ may be mediated
through regulation of BDNF (Chuang et al., 2002;Jope, 1999;Manji and Duman, 2001). BDNF is one of the growth factors called neurotrophins (NTs), and it regulates the survival, differentiation, and function of neurons. The effect of BDNF is mediated by the tyrosine kinase receptor of the Trk B. BDNF binds to TrkB and initiates a number of anti-apoptotic signaling pathways, including phosphatidylinositol 3-kinase (PI3K)/Akt pathway (Ho et al., 2002;Jaboin et al., 2002;Patapoutian and Reichardt, 2001).
Recently, it has been discovered that Li+ selectively protects spinal motor neurons in
animal models of amyotrophic lateral sclerosis (ALS) by preventing the apoptosis component (Shin et al., 2007). ALS is a neurodegenerative disorder characterized by degeneration of upper and lower motor neurons, progressive paralysis, and an average mortality of 5 years after onset.
It has been reported that the SN showed mild degeneration and Lewy bodies, known for presymptomatic Parkinson’s disease, in an autopsy of an ALS patient’s SN (Andreassen et al.,
2001a;Su et al., 1996).Andreassen et al. (2001) reported that TH neurons decreased in the
2
(SOD-1), causing familial ALS (Andreassen et al., 2001b;Andreassen et al., 2001c). However, the reason why degeneration of TH neurons occurs in ALS remains an unanswered question.
TrkB is expressed in SN neurons (Benisty et al., 1998;Nishio et al., 1998). The activation of TrkB is shown to promote the survival of SN neurons and protect them against excitotoxic insult (Hynes et al., 1994;Volpe et al., 1998).
The present study hypothesizes that Li+ would probably have effects on neuronal survival.
For the pharmacological prevention of apoptosis, Li+, a well known anti-apoptotic agent, was
3
Ⅱ. MATERIAL AND METHODS
A. Materials
1. Experimental Animal Models
G93A mice and control littermate mice, aged 8 weeks, were used, and kept in conditions of constant temperature and humidity. Mice were fed with regular rodent chow (control) or a 0.2% lithium carbonate (Li+)-containing diet for 4-8weeks. A bottle of saline was available
for the rats and mice. Blood samples for monitoring the Li+ concentration were collected.
B. Methods
1. SYBR Green I reverse transcription-polymerase chain reaction
The cerebral cortex, hippocampus formation, striatum, midbrain, and cerebellum were removed, frozen in dry ice, and stored at -80℃. For RT-PCR, cDNA was synthesized in 20㎕ reaction buffer containing 4㎕5×M-MLV buffer, 0.25㎕ M-MLV RTase (200 U/ml; TaKaRa, Takara, Japan), 0.25 ㎕ RNase inhibitor (40 U/ml; TaKaRa), 8㎕ 2.5mM dNTP, 3.5㎕ oligo dT and 2㎕ template RNA. The thermal profile for RT was 42ºC for 30 min and 95ºC
for 5 min. The PCR was carried out in a reaction volume of 20㎕ containing10㎕ SYBR
Green PCR kit (Qiagen), 2㎕ template cDNA, and forward and reverse primers 0.5㎕ (10pmole). The thermal profile for PCR was 95ºC for 10 min, followed by 40 cycles of 95ºC
4
for 10 s, and 56ºC for 15 s, and 72ºC for 20s. The Ct used in the real-time PCR
quantification was defined as the PCR cycle number that crossed an arbitrarily chosen signal threshold in the log phase of the amplification curve. After the PCR, data were viewed and analyzed using the Rotor Gene RG-3000 software (Corbett, RG-3000, Australia). The actual sequences of specific primers were as follows: BDNF (forward) 5’-GGTCACAGTCCTGGAGAAAG-3’, (reverse) 5’-GTCTATCCTTATGAACCGCC-3; GAPDH(forward) TCCCTCAAGATTGTCAGCAA-3’; (reverse) 5’-AGATCCACAACGGATACATT-3’
2. Stereology
Cryostat sections were cut at 40㎛ through the whole substantia nigra (SN) from bregma -2.70 to -3.80. Every seventh slide was immunostained for tyrosine hydroxylase (TH) (Chemicon) and demonstrated using goat anti-rabbit biotinylated secondary antibody (Vector Laboratories), followed by peroxidase-diaminobenzidine standard Vectastin Elite ABC kit (Vector Laboratories). To estimate the total number of TH positive neuronal cells in the SN the optical fractionator method was used. Areas were outlined using a 4X objective and counts performed using a 100X oil immersion objective. For quantification, the Visiopharm Integrator System (VIS, new CAST) software (Visiopharm, Denmark) was used on a PC system connected to an Olympus BX-51 microscope. The total number of cells (N) in the SN was estimated by using the equation:
5
Where ∑Q is the total number of TH positive cells counted, hsf is the thickness of the sampling fraction, asf is the area of the sampling fraction, and ssf is the section of the sampling fraction.
3. Immuohistochemistry
Brain sections were fixed in 3% paraformaldehyde, washed in PBS, incubated in 0.3%
H2O2 and 0.25% Triton X-100 for 10 min at room temperature, and reacted with 10% horse
serum for 1 h. Sections were then reacted overnight at 4℃ with the primary anti-nitrotyrosine (4μg/ml, Upstate), NeuN (Chemicon), cleaved-caspase 8 and cleaved-caspase 9 (cellsignaling). Next, the sections were reacted with anti-rabbit biotin-conjugated antibody, anti-rabbit rhodamin-conjugated antibody and anti-mouse fluorescence-conjugated antibody (Vector laboratories) for 2 h. The biotin-labeled sections were incubated with avidin-biotin-peroxidase complex (Vector laboratories) for 1 h and then visualized using 3,3’-diaminobenzidine tetrahydrochloride dehydrate (DAB).
4. Fluorescence in situ hybridization
Brains were removed after the mice were sacrificed. A coronal brain section (20㎛ thick) was cut with a cryostat and mounted onto gelatin-coated slides. Fluorescence in situ hybridization (FISH) was performed with a digoxingenin (DIG)-UTP-labeled cRNA probe. Plasmid pBS contained a BDNF cDNA fragment (750 bp). This plasmid was linearized, and the DIG-labeled cRNA probe was prepared by in vitro transcription using T3 RNA polymerase (antisense), and T7 RNA polymerase (sense) as a control. All prehybridization
6
procedures were performed under RNase-free conditions at room temperature. Hybridization was carried out at 52℃ overnight in a hybridization buffer (50% deionized formamide, 20X SSC, 50X Denhardt’s solution, 50% dextran sulfate,10mg/ml yeast tRNA, and 10mg/ml salmon sperm DNA). After hybridization, the sections were washed with 2× SSC and then treated with RNase A (100ng/ml) at 37℃ for 30 min. After thorough washing, the sections were incubated with anti-digoxigenin-rhodamine (diluted 1:1000, Roche) for 2hr at room temperature. The substantia nigra (SN) in the midbrain was analyzed.
5. Fluorescent intensity
Intensity (Fi) was estimated for each slice by acquiring intensity values in nine different areas within the SN. Simultaneously, the average background intensity (F0) was obtained for
each slice in three different areas. The average intensity (Fm) was then calculated (Fm=Fi-F0). (MetaMorph was the analyzing program used.)
6. Data analysis
Results of experiments are expressed as the mean±S.E.M. Experiments containing two independent groups of mice were subjected to the independent-samples t-test. Analysis of variance and the Student Neuman-Keuls test were used for multiple comparisons. Statistical significance was set at P < 0.05. (n=5)
7
Ⅲ. RESULTS
A. Lithium increased BDNF mRNA expression after chronic treatment in brain and spinal cord.
Li+ concentration in plasma
The Li+ concentration was 0.48±0.02 mmol/L (n=9). In humans, the Li+ therapeutic
concentration range is 0.8~1.2 mmol/L.
Li+ increased BDNF mRNA expression in brain and spinal cord.
The major aim of this study was to carry out a detailed analysis of BDNF mRNA levels
following chronic Li+ treatment. BDNF mRNA expression was determined by SYBR
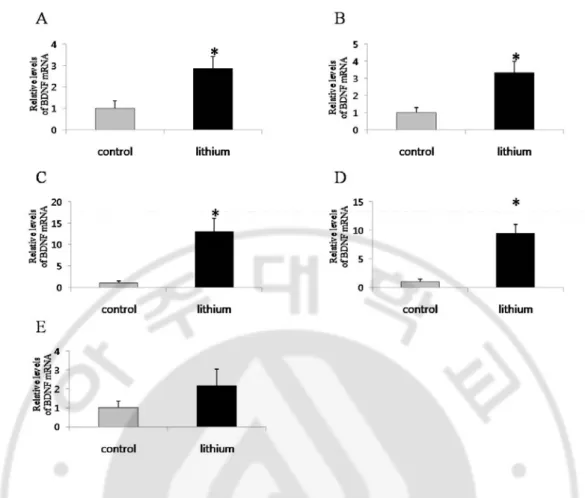
real-time PCR. Li+ treatment increased BDNF mRNA in the cerebral cortex (Fig. 1A),
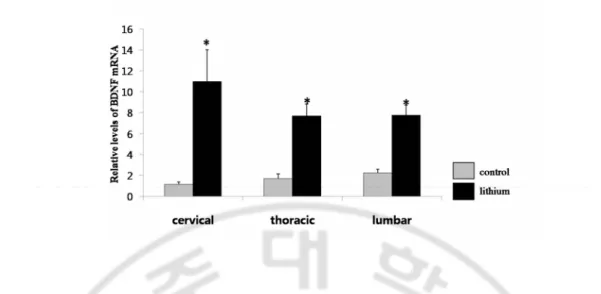
hippocampal formation (Fig. 1B), striatum (Fig. 1C), midbrain (Fig. 1D), but not in the cerebellum (Fig. 1E). The BDNF mRNA expression increased in the spinal cord (divided
into three parts: cervical, thoracic, and lumbar) following Li+ treatment (Fig. 2). These
results mean that chronic Li+ treatment increases BDNF expression level in most of the CNS
8
Fig. 1. Li+ increases BDNF mRNA expression in various brain areas in adult rat. Real Time PCR analysis of BDNF mRNA in the cerebral cortex (A), hippocampal formation (B), striatum (C), midbrain (D), and cerebellum (E) in control rats and rats treated with Li+ for 3
9
Fig. 2. Li+ increases expression of BDNF mRNA in the spinal cord of adult rat. Real Time PCR analysis of BDNF mRNA in the cervical, thoracic, and lumbar spinal cord of adult rat treated with Li+ for 3 weeks , mean ±S.E.M.(n = 5~9), *, significant differences
10
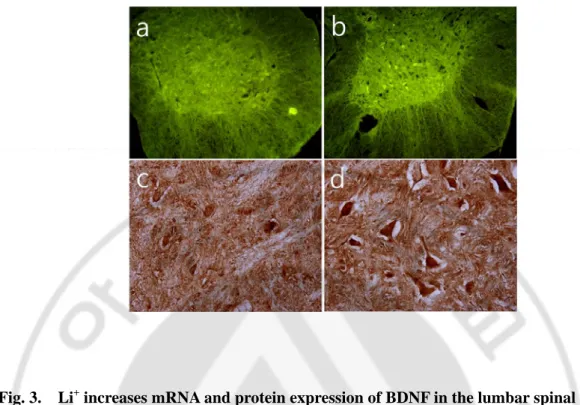
mRNA and protein of BDNF were exclusively localized in ventral motor neurons of lumbar segment in a lithium-sensitive manner.
Whether chronic Li+ treatment would have an influence on the expression of BDNF
mRNA (Fig 3, a and b) and the protein of BDNF (Fig 3, c and d) was examined. mRNA and the protein of BDNF were selectively expressed in the motor neuron of the ventral horn in
the mouse treated with Li+. These results indicate that mRNA and the protein of BDNF were
exclusively localized in ventral motor neurons of lumbar segments in a lithium-sensitive manner.
B. Li+ had an effect on the protection neurons from degenerative death in the ALS animal model
Li+ attenuated degeneration of spinal motor neurons in the ALS animal model.
Next, it was examined whether protection from neuronal death in neurodegenerative diseases could be exerted by increased BDNF due to chronic Li+ treatment. ALS is one of the
neurodegenerative diseases in which motor neurons die progressively and skeletal muscles
denervate. ALS animal models, G93A mice, 16-weeks-old, fed chow containing Li+
(~200mg/kg/d) for three weeks were used. Then the lumbar regions of the spinal cords were obtained and stained with cresyl violet (Fig. 4A) and MAP-2/ChAT (Fig. 4B). In the G93A mice (Fig. 4A-b and B-b), most motor neurons died and axons degenerated, but in the G93A mouse group treated with lithium (Fig. 4A-c and B-c) motor neurons and axons were protected from degenerative death. The BDNF protein level and its downstream signal,
11
phosphorylation of Akt (Ser473 and Thr308), were examined to confirm that BDNF was
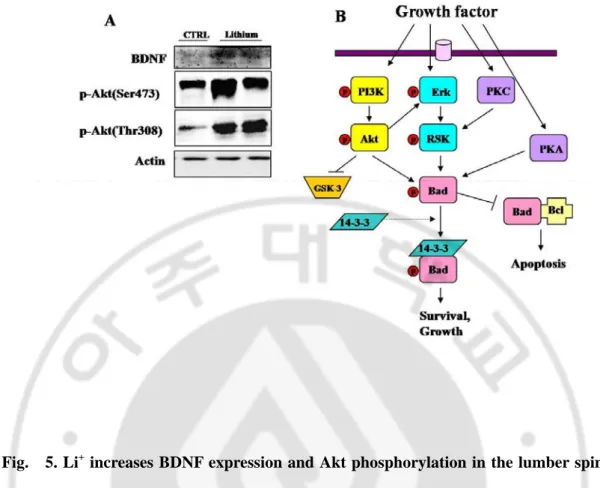
responsible for this protection. As can be seen in Fig. 5, Li+ increased the BDNF protein
level as well as the BDNF mRNA level. Also, increased BDNF started the survival signaling pathway as indicated by the increasing phosphorylation of Akt.
In the ALS animal model, the number of TH positive cells in the SN was decreased. It has been reported that the cell numbers of dopaminergic neurons in the SN of
transgenic ALS mice decreased. To examine if Li+ would protect the dopaminergic neurons
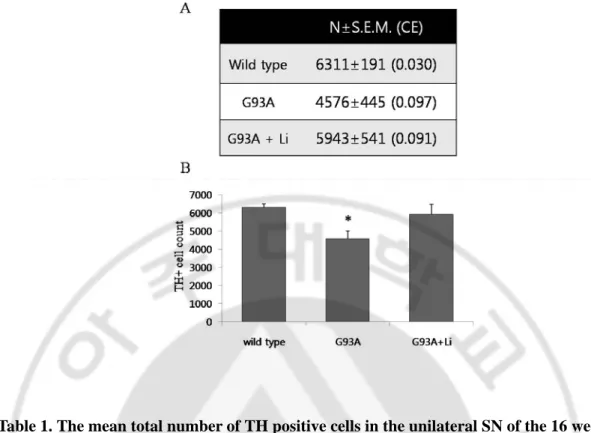
in ALS mice, loss of the dopaminergic neurons was first verified by counting cells immunoreactive to tyrosine hydroxylase (TH) antibody using a stereology system (CAST, Visopharm) method in 16-week old male G93A mice (Table 1). The mean total number of TH positive cells in the unilateral SN of the mice was estimated to be 4,500 (coefficient of variation (CV)=0.22) (with a mean coefficient of error (CE) of 0.09). It decreased as much as 28% from that of the littermate control mice (Table 1). In this study, the CE2/CV2 ratio is 0.2,
verifying a low methodological bias in the precision of the estimated neuronal number (West, 1999). In ALS, the progress of cell death occurs in dopaminergic neurons in the SN as well as in spinal motor neurons. In addition, it is presumed that Li+ increased the BDNF level in
12
Fig. 3. Li+ increases mRNA and protein expression of BDNF in the lumbar spinal motor neurons of adult rat. Fluorescence (a and b, in situ hybridization with anti-sense BDNF cRNA probe ) and bright-field (c and d, immunocytochemistry with a BDNF antibody) photomicrographs of the lumbar ventral sections in control rats (a and c) and rats treated with Li+ for 3 weeks (b and d).
13
Fig. 4. Li+ attenuates degeneration of spinal motor neurons in ALS mice. (A) Bright-field (A, cresyl vilotet staining) and fluorescence (B, double immunocytochemistry of MAP-2 (green) and ChAT (red) antibodies] in 16 week-old control littermate mouse (a), G93A
transgenic mice (b), and G93A transgenic mouse treated with Li+ (~200 mg/kg/d) starting
from 8 weeks of age (c).
14
Fig. 5. Li+ increases BDNF expression and Akt phosphorylation in the lumber spinal cord of adult mice. (A) Western blot analysis of BDNF, p-Akt(Ser473), and p-Akt(Thr308)
in the lumbar spinal cord of B6SJL mice treated with vehicle control (CTRL) or Li+ for 3
15
Table 1. The mean total number of TH positive cells in the unilateral SN of the 16 weeks old male G93A mouse. Table (A), Histogram (B). mean ±S.E.M.(n = 5), *, significant differences compared with control, p<0.05.
16 Li+ prevents apoptotic cell death.
In this study, the mechanisms that could influence the protection of dopaminergic neurons after chronic lithium administration were investigated. Cell death due to apoptosis was first determined in the male 16 week-old G93A mice. Apoptotic signals are caspase-9 (Fig. 6A, wild type 100±50.077%, G93A 382.40±64.091%, G93A+Li 145.94±10.985%), caspase-8 (Fig. 6B, wild type 100±33.823%, G93A 1411.08±465.512%, G93A+Li 633.63±192.976%), and caspase-3 (Fig. 6C, wild type 100±82.456%, G93A 697.25±146.999%, G93A+Li 154.26±94.755%). These signals were increased in the transgenic mice but Li+ decreased the apoptotic signals (Fig. 6D). This could be one of the
modes of neuron cell death protection by Li+ in the SN.
Li+ cannot reduce the ROS level.
It was then investigated as to whether lithium could inhibit oxidative stress (Fig. 7). In the familiar ALS animal model, lower motor neurons died from oxidative stress. Immunohistochemistry with nitrotyrosine antibody staining was used to see if dopaminergic neuronal death in the SN of the 12 week-old male mouse is involved with oxidative stress, and if Li+ reduces the ROS level. The result proved that the ROS level was increased in the
G93A mouse, and that lithium (administration for 28 days) did not reduce the ROS level. This means that lithium did not provide protection against neuronal cell death through the ROS inhibition mechanism. The protection effect was attributed to the anti-apoptotic mechanism.
19
Fig. 6 . Li+ prevents apoptotic neuronal cell death. (A) Fluorescence photomicrographs (a, e,and i, immunocytochemistry of caspase-9; b,f, and j, immunocytochemistry of NeuN; c,g, and k, DAPI ; d, h and l, triple merge ) in 16 week-old control littermate mouse (a, b, c and d), G93A transgenic mice (e, f, g and h), and G93A transgenic mouse treated with Li+ (~200
mg/kg/d) starting from 8 weeks of age (I, j, k and l). (B) Fluorescence photomicrographs (a, e,and i, immunocytochemistry of caspase-8; b,f, and j, immunocytochemistry of NeuN; c,g, and k, DAPI ; d, h and l, triple merge ) in 16 week-old control littermate mouse (a, b, c and d), G93A transgenic mice (e, f, g and h), and G93A transgenic mouse treated with Li+ (~200
mg/kg/d) starting from 8 weeks of age (I, j, k and l). (C) Fluorescence photomicrographs (a, e,and i, immunocytochemistry of caspase-3; b,f, and j, immunocytochemistry of NeuN; c,g, and k, DAPI ; d, h and l, triple merge ) in 16 week-old control littermate mouse (a, b, c and d), G93A transgenic mice (e, f, g and h), and G93A transgenic mouse treated with Li+ (~200
mg/kg/d) starting from 8 weeks of age (I, j, k and l). (D) Fluorescence intensity quantitation of caspase-9 (a), -8 (b), and-3 (c). mean ±S.E.M.(n = 5~9), *, significant differences compared with control, p<0.05.
20
Fig. 7. Li+ cannot reduce ROS level and cannot prevent ROS-mediated cell death. Bright-field photomicrographs (immunocytochemistry of nitrotyrosine) in 12 week-old control littermate mouse (a), G93A transgenic mice (b), and G93A transgenic mouse treated with Li+ (~200 mg/kg/d) starting from 8 weeks of age (c).
21 Li+ increases BDNF mRNA level
Chronic lithium treatment increase BDNF expression. Our animal model increased BDNF mRNA level also. We carried out fluorescence in situ hybridization (FISH) for BDNF mRNA expression (Fig. 8), lithium treatment group (for 28 days) was increased fluorescent intensity about 44% more than transgenic G93A group (wild type, 100±12.548; wild type+
Li+ ,192.38±11.612; G93A, 62.63±2.975; G93A+ Li+ ,97.47±11.065%). BDNF plays a
central role in cell survival and activity dependent neural plasticity. This increasing BDNF level might exert protecting dopaminergic neurons from dying.
23
Fig. 8. Li+ increases expression of BDNF mRNA in the substantia nigra in ALS animal model. (A) Fluorescence photomicrographs (a, e,and i, in situ hybridization of BDNF mRNA; b,f, and j, immunocytochemistry of TH; c,g, and k, DAPI ; d, h and l, triple merge ) in 12 week-old control littermate mouse (a, b, c and d), control littermate mouse treated with Li+ (~200 mg/kg/d) starting from 8 weeks of age (e, f, g and h), G93A transgenic mouse (I, j,
k and l). and G93A transgenic mouse treated with Li+ (m,n,o and p). (B) Fluorescence
intensity quantitation of BDNF mRNA. mean ±S.E.M.(n = 5~9), *, significant differences compared with control, p<0.05.
24
Ⅳ. DISCUSSION
In the present study, treatment with Li+ for 21 days increased the BDNF expression in the
cerebral cortex, hippocampal formation, striatum, midbrain, and spinal cord of the test subjects. However, acute treatment did not increase BDNF expression in any of the regions examined (data not shown). These results indicate that therapeutically more relevant, chronic
treatment with Li+ enhances BDNF expression even at subtherapeutic concentrations
(average 0.48mM). A variety of antidepressant modalities, when administered chronically, are known to have the characteristic neuroplastic effect of upregulation of BDNF in the hippocampus and frontal cortex (Duman, 2002;Nibuya et al., 1995). The present study indicates that Li+ shares the same neuroplastic effect as antidepressants. In addition, chronic
administration of Li+ upregulates BDNF expression in other brain regions that are involved
in neurodegenerative diseases.
It is suggested that Li+ may exert neuroprotective actions by increasing BDNF expression
in specific regions of the CNS. However, our result seems to be inconsistent with other studies showing decreased BDNF level in the frontal cortex (Angelucci et al., 2003) by long term treatment with Li+.
There have been studies demonstrating increase in BDNF expression in the hippocampus
by chronic Li+ treatment in animals (Drevets, 1999;Einat et al., 2003;Fukumoto et al.,
2001;Jacobsen and Mork, 2004).
There has been evidence of enhancement of synaptic plasticity by lithium as evidenced by increase in long term potentiation (LTP) in the hippocampal subregions by subchronic (Shim et al., 2007) and chronic lithium administration (Son et al., 2003). Son et al (2003)
25
reported the enhancement of LTP and the concomitant increase in BDNF level in the dentate gyrus (DG) after a 4- week Li+ treatment. These findings, plus the present results, suggest
that an increase in BDNF expression may contribute to Li+-induced enhancement of synaptic
plasticity.
Pharmacological activation of the cAMP-CREB pathway leads to an increase in hippocampal neurogenesis (Nakagawa et al., 2002;Williams et al., 2001;Zhu et al., 2004). It has been demonstrated that chronic Li+ treatment increases neurogenesis in the hippocampus
with accompanying increases in cyptoprotective or neurotrophic molecules such as Bcl-2 and BDNF (Chen et al., 2000;Son et al., 2003).
The detailed mechanism of upregulation of BDNF expression in the specific brain regions is not fully understood. However, there are observations that cAMP responsive
element binding protein (CREB) activity is modulated by Li+. The CREB binding activity
and phosphorylated CREB levels are enhanced by the therapeutically relevant concentration of Li+ in cultured cerebellar granule cells (Ozaki and Chuang, 1997) and in the brain of the
rat (Ozaki and Chuang, 1997). Phosphorylated CREB levels are increased in the rat hippocampus and frontal cortex by chronic Li+ administration (Einat et al., 2003). CREB is a
common target of diverse signal transduction pathways including Ca2+
-calmodulin-dependent kinase, protein kinase C, ribosomal S6 kinase, and protein kinase A (Duman,
2002;Finkbeiner, 2000), and BDNF expression is modulated by CREB activated by the Ca2+
signal (Shieh and Ghosh, 1999).
This result means that Li+ exerts protection from neuronal cell death. In nitrotyrosine
immunohistochemistry, the ROS level did not differ between G93A mice and G93A mice with Li+ treatment, and in both groups the ROS level was higher than the control littermate
26
group. But in the caspase-9, 8 and 3 immunohistochemistry of 16-week old G93A mice, the fluorescent intensity of the Li+ administration group was lower than its G93A group. These
data show that Li+ can protect neurons from apoptosis but it cannot provide protection from
oxidative stress. Therefore, Li+ protects neurons of the SN through the mechanism of the
27
Ⅴ. CONCLUSION
1. Chronic (3 weeks) administration of Li+ increases mRNA expression of BDNF in
the brain and the spinal cord in adult rat.
2. Chronic administration of Li+ induces BDNF expression and AKT phosphorylation
in the spinal cord of adult mouse.
3. Chronic administration of Li+ protects motor neurons in spinal cord and
doparminergic neurons in SN from neurodegenerative cell death.
4. These protecting effects engage increased BDNF level by Li+ treatment, block the
apoptotic signals.
The present study suggests that chronic treatment with Li+ promotes neuronal survival
and regeneration through upregulation of BDNF in certain brain areas (e.g. SN) including the lumbar spinal motor neurons.
28
REFERENCES
1. Andreassen, O. A., R. J. Ferrante, P. Klivenyi, A. M. Klein, A. Dedeoglu, D. S. Albers, N. W. Kowall and M. F. Beal, Transgenic ALS mice show increased vulnerability to the mitochondrial toxins MPTP and 3-nitropropionic acid, Exp.
Neurol., 168(2), 356-363, 2001a.
2. Andreassen, O. A., R. J. Ferrante, P. Klivenyi, A. M. Klein, A. Dedeoglu, D. S. Albers, N. W. Kowall and M. F. Beal, Transgenic ALS mice show increased vulnerability to the mitochondrial toxins MPTP and 3-nitropropionic acid, Exp.
Neurol., 168(2), 356-363, 2001b.
3. Andreassen, O. A., R. J. Ferrante, P. Klivenyi, A. M. Klein, A. Dedeoglu, D. S. Albers, N. W. Kowall and M. F. Beal, Transgenic ALS mice show increased vulnerability to the mitochondrial toxins MPTP and 3-nitropropionic acid, Exp.
Neurol., 168(2), 356-363, 2001c.
4. Angelucci, F., L. Aloe, P. Jimenez-Vasquez and A. A. Mathe, Lithium treatment alters brain concentrations of nerve growth factor, brain-derived neurotrophic factor and glial cell line-derived neurotrophic factor in a rat model of depression, Int. J.
29
5. Benisty, S., F. Boissiere, B. Faucheux, Y. Agid and E. C. Hirsch, trkB messenger RNA expression in normal human brain and in the substantia nigra of parkinsonian patients: an in situ hybridization study, Neuroscience, 86(3), 813-826, 1998.
6. Chen, G., G. Rajkowska, F. Du, N. Seraji-Bozorgzad and H. K. Manji, Enhancement of hippocampal neurogenesis by lithium, J. Neurochem., 75(4), 1729-1734, 2000.
7. Chuang, D. M., R. W. Chen, E. Chalecka-Franaszek, M. Ren, R. Hashimoto, V. Senatorov, H. Kanai, C. Hough, T. Hiroi and P. Leeds, Neuroprotective effects of lithium in cultured cells and animal models of diseases, Bipolar. Disord., 4(2), 129-136, 2002.
8. Drevets, W. C., Prefrontal cortical-amygdalar metabolism in major depression, Ann.
N. Y. Acad. Sci., 877, 614-637, 1999.
9. Duman, R. S., Pathophysiology of depression: the concept of synaptic plasticity,
Eur. Psychiatry, 17 Suppl 3, 306-310, 2002.
10. Einat, H., P. Yuan, T. D. Gould, J. Li, J. Du, L. Zhang, H. K. Manji and G. Chen, The role of the extracellular signal-regulated kinase signaling pathway in mood modulation, J. Neurosci., 23(19), 7311-7316, 2003.
30
11. Finkbeiner, S., Calcium regulation of the brain-derived neurotrophic factor gene,
Cell Mol. Life Sci., 57(3), 394-401, 2000.
12. Fukumoto, T., S. Morinobu, Y. Okamoto, A. Kagaya and S. Yamawaki, Chronic lithium treatment increases the expression of brain-derived neurotrophic factor in the rat brain, Psychopharmacology (Berl), 158(1), 100-106, 2001.
13. Ho, R., A. Eggert, T. Hishiki, J. E. Minturn, N. Ikegaki, P. Foster, A. M. Camoratto, A. E. Evans and G. M. Brodeur, Resistance to chemotherapy mediated by TrkB in neuroblastomas, Cancer Res., 62(22), 6462-6466, 2002.
14. Hynes, M. A., K. Poulsen, M. Armanini, L. Berkemeier, H. Phillips and A. Rosenthal, Neurotrophin-4/5 is a survival factor for embryonic midbrain dopaminergic neurons in enriched cultures, J. Neurosci. Res., 37(1), 144-154, 1994.
15. Jaboin, J., C. J. Kim, D. R. Kaplan and C. J. Thiele, Brain-derived neurotrophic factor activation of TrkB protects neuroblastoma cells from chemotherapy-induced apoptosis via phosphatidylinositol 3'-kinase pathway, Cancer Res., 62(22), 6756-6763, 2002.
16. Jacobsen, J. P. and A. Mork, The effect of escitalopram, desipramine, electroconvulsive seizures and lithium on brain-derived neurotrophic factor mRNA and protein expression in the rat brain and the correlation to 5-HT and 5-HIAA levels,
31 Brain Res., 1024(1-2), 183-192, 2004.
17. Jope, R. S., Anti-bipolar therapy: mechanism of action of lithium, Mol. Psychiatry,
4(2), 117-128, 1999.
18. Kang, H. J., J. S. Noh, Y. S. Bae and B. J. Gwag, Calcium-dependent prevention of neuronal apoptosis by lithium ion: essential role of phosphoinositide 3-kinase and phospholipase Cgamma, Mol. Pharmacol., 64(2), 228-234, 2003.
19. Manji, H. K. and R. S. Duman, Impairments of neuroplasticity and cellular resilience in severe mood disorders: implications for the development of novel therapeutics, Psychopharmacol. Bull., 35(2), 5-49, 2001.
20. Nakagawa, S., J. E. Kim, R. Lee, J. E. Malberg, J. Chen, C. Steffen, Y. J. Zhang, E. J. Nestler and R. S. Duman, Regulation of neurogenesis in adult mouse hippocampus by cAMP and the cAMP response element-binding protein, J. Neurosci., 22(9), 3673-3682, 2002.
21. Nibuya, M., S. Morinobu and R. S. Duman, Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments,
J. Neurosci., 15(11), 7539-7547, 1995.
32
motoneurons of amyotrophic lateral sclerosis, Neuroreport, 9(7), 1661-1665, 1998.
23. Ozaki, N. and D. M. Chuang, Lithium increases transcription factor binding to AP-1 and cyclic AMP-responsive element in cultured neurons and rat brain, J.
Neurochem., 69(6), 2336-2344, 1997.
24. Patapoutian, A. and L. F. Reichardt, Trk receptors: mediators of neurotrophin action, Curr. Opin. Neurobiol., 11(3), 272-280, 2001.
25. Shieh, P. B. and A. Ghosh, Molecular mechanisms underlying activity-dependent regulation of BDNF expression, J. Neurobiol., 41(1), 127-134, 1999.
26. Shim, S. S., M. D. Hammonds, S. J. Ganocy and J. R. Calabrese, Effects of sub-chronic lithium treatment on synaptic plasticity in the dentate gyrus of rat hippocampal slices, Prog. Neuropsychopharmacol. Biol. Psychiatry, 31(2), 343-347, 2007.
27. Shin, J. H., S. I. Cho, H. R. Lim, J. K. Lee, Y. A. Lee, J. S. Noh, I. S. Joo, K. W. Kim and B. J. Gwag, Concurrent administration of Neu2000 and lithium produces marked improvement of motor neuron survival, motor function, and mortality in a mouse model of amyotrophic lateral sclerosis, Mol. Pharmacol., 71(4), 965-975, 2007.
33
28. Son, H., I. T. Yu, S. J. Hwang, J. S. Kim, S. H. Lee, Y. S. Lee and B. K. Kaang, Lithium enhances long-term potentiation independently of hippocampal neurogenesis in the rat dentate gyrus, J. Neurochem., 85(4), 872-881, 2003.
29. Su, M., K. Wakabayashi, Y. Tanno, T. Inuzuka and H. Takahashi, [An autopsy case of amyotrophic lateral sclerosis with concomitant Alzheimer's and incidental Lewy body diseases], No To Shinkei, 48(10), 931-936, 1996.
30. Volpe, B. T., J. Wildmann and C. A. Altar, Brain-derived neurotrophic factor prevents the loss of nigral neurons induced by excitotoxic striatal-pallidal lesions,
Neuroscience, 83(3), 741-748, 1998.
31. West, M. J., Stereological methods for estimating the total number of neurons and synapses: issues of precision and bias, Trends Neurosci., 22(2), 51-61, 1999.
32. Williams, B. M., Y. Luo, C. Ward, K. Redd, R. Gibson, S. A. Kuczaj and J. G. McCoy, Environmental enrichment: effects on spatial memory and hippocampal CREB immunoreactivity, Physiol Behav., 73(4), 649-658, 2001.
33. Zhu, D. Y., L. Lau, S. H. Liu, J. S. Wei and Y. M. Lu, Activation of cAMP-response-element-binding protein (CREB) after focal cerebral ischemia stimulates neurogenesis in the adult dentate gyrus, Proc. Natl. Acad. Sci. U. S. A, 101(25), 9453-9457, 2004.
34 –국문요약–
장기간의 리튬 투여와 특정 뇌 영역의 신경영양인자 (Brain Derived
Neurotrophic Factor (BDNF) )mRNA의 증가
아주대학교 대학원 신경과학기술 협동과정 (지도교수 : 곽병주) 우울증 치료제로 이용되는 리튬은 세포고사에서 세포를 보호하는 효과는 있으나 산화 적 손상으로부터 세포를 보호하지는 못한다. 앞선 연구에서 G93A 동물에서 리튬이 생존 기간과 운동 기능을 현저하게 향상시키는 결과를 보고한바있으며, ALS 환자에서 흑질부 위의 도파민 신경세포의 숫자가 감소한다는 결과가 알려져있다. 파킨슨 병에서 흑질의 도파민 신경세포가 취약하다는 것 역시 잘 알려져 있다. 이 연구에서 ALS 동물 모델의 흑질 부위의 도파민 신경세포의 수를 측정한 결과 대조군에 비해 세포의 수가 20% 정도 감소하였으며, 리튬을 투여한 그룹에서는 세포의 손실이 보이지 않았다. 이 결과는 리튬 이 신경세포의 사멸을 막는 역할을 하고 있음을 말한다. 리튬은 세포고사의 기전인 caspase의 활성을 막음으로서 ALS동물 모델의 도파민 신경세포와 운동 신경세포를 신경 세포사로 부터 보호한다. Nitrotyrosine을 이용한 활성화산소 증감정도를 측정한 실험에 서 리튬을 처리한 군에서 ROS가 감소하지 않는 것으로 보아 리튬의 세포 보호 효과는 ROS를 막는 기전으로 일어나지 않음을 알 수 있었다. 이러한 신경세포 보호기전의 근본 은 리튬의 장기 투여로 인한 BDNF의 증가에 따른 것으로 보여진다. 핵심단어 : BDNF, ALS, G93A, ROS, Apoptosis


![Fig. 4. Li + attenuates degeneration of spinal motor neurons in ALS mice. (A) Bright- Bright-field (A, cresyl vilotet staining) and fluorescence (B, double immunocytochemistry of MAP-2 (green) and ChAT (red) antibodies] in 16 week-old control littermate](https://thumb-ap.123doks.com/thumbv2/123dokinfo/4677026.1625/24.892.147.753.153.468/attenuates-degeneration-bright-staining-fluorescence-immunocytochemistry-antibodies-littermate.webp)