하시모토병과 동반된 진성적혈구무형성증 1예
경상대학교 의과대학 내과학교실, 1분당서울대학교병원 내과
김훈구ㆍ이경원ㆍ김광민ㆍ강명희ㆍ주지현ㆍ강정훈ㆍ이종석
1
A Case of Pure Red Cell Aplasia Associated with Hashimoto Disease
Hoon-Gu Kim, M.D., Gyeong-Won Lee, M.D., Kwang-Min Kim, M.D., Myung-Hee Kang, M.D., Ji-Hyun Ju, M.D., Jung Hun Kang, M.D. and Jong-Seok Lee, M.D.1
Department of Internal Medicine, Gyeongsang National University School of Medicine, Jinju,
1Seoul National University Bundang Hospital, Seongnam, Korea
Pure red cell aplasia (PRCA) is a rare hematologic disorder characterized by anemia, reticulocytopenia in the blood and isolated severe erythroblastopenia with no abnormalities of the granulopoiesis and mega- karyopoiesis in the bone marrow. Acquired PRCA has been reported in association with various auto- immune diseases, such as systemic lupus erythematosus (SLE), rheumatoid arthritis, mixed connective tissue disease and Sjögren's syndrome. We report here on a case of PRCA associated with Hashimoto disease (without any other autoimmune disease) which, to the best of our knowledge, has not been pre- viously reported in Korean and English literatures. Our patient was treated with levothyroxine alone with- out other immunosuppressive agents and her hemoglobin concentration and hematocrit values returned to normal. (Korean J Hematol 2007;42:146-150.)
Key Words: Pure red cell aplasia, Hashimoto disease, Autoimmune, Hypothyroidism
146
Correspondence to:Gyeong-Won Lee, M.D.
Department of Internal Medicine, Gyeongsang National Univer- sity School of Medicine
92, Chilam-dong, Jinju 660-702, Korea Tel: +82-55-750-8066, Fax: +82-55-758-9122 E-mail: [email protected]
접수:2007년 2월 26일, 수정:2007년 4월 17일 승인:2007년 4월 18일
교신저자:이경원, 경남 진주시 칠암동 92
660-702, 경상대학교 의과대학 내과학교실 Tel: 055-750-8066, Fax: 055-758-9122
E-mail: [email protected] 서 론
진성적혈구무형성증(pure red cell aplasia, PRCA)은 골수에서 백혈구계과 거핵구계의 형성은 정상이면서 적혈구계의 선택적인 생성부전으로 인하여 빈혈, 망상 적혈구감소증과 골수 내 적아구의 저형성증을 특징으 로 하는 드문 질환이다. 진성적혈구무형성증의 원인으 로 후천적인 경우 흉선종 외에도 감염, 약제, 자가면역 질환 등이 알려져 있다.1)
진성적혈구무형성증과 관련된 면역질환으로 전신홍
반루프스(systemic lupus erythematosus, SLE), 류마티 스관절염,2) 혼합결합조직병(mixed connective tissue disease),3) 쇼그렌증후군4) 등이 알려져 있지만, 진성적 혈구무형성증이 이러한 다른 자가면역질환의 동반 없 이 하시모토병(Hashimoto disease)과 관련되어 발생된 예는 국내 및 영문 문헌 고찰상 보고된 바가 없다.
이에 저자들은 62세 여자 환자에서 하시모토병으로 인한 갑상선기능저하증과 동반된 진성적혈구무형성증 을 경험하였기에 보고하는 바이다.
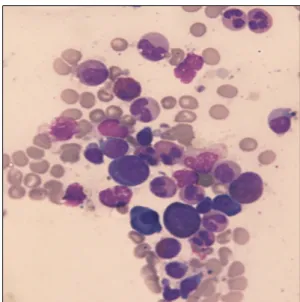
Fig. 1. Bone marrow aspirate smear showing marked de- crease of erythroid precursors (Wright-Giemsa stain, ×400).
증 례
환 자: 안○○, 62세, 여자 주 소: 현기증
현병력: 약 한 달 전부터 시작된 현기증을 주소로 개 인 병원에 내원하여 시행한 혈액검사상 빈혈이 발견되 어 본 병원으로 전원되었다. 본원 내과에 입원하여 갑 상선기능저하증 및 빈혈로 진단 받고 입원 5일째부터 levothyroxine을 투여하기 시작하여 일단 외래 경과를 관찰하기로 하고 입원 9일째 퇴원하였다. 환자는 퇴원 후 매일 levothyroxine 100㎍을 복용하며 지내다 퇴원 약 한 달 후 외래에서 시행한 혈액검사상 빈혈이 더욱 악화된 소견을 보여 정밀 검사를 위해 다시 입원하였다.
과거력: 특이사항은 없었다.
가족력: 특이사항은 없었다.
이학적 소견: 재입원 당시 체온 36.7oC, 맥박수 분당 82회, 호흡수 분당 20회, 혈압 110/70mmHg이었고, 안 면은 창백하였으며 공막의 황달 소견은 보이지 않았고 결막에 빈혈상을 보였다. 경부에서 미만성의 갑상선 종대가 있었으며, 촉진되는 림프절은 없었다. 흉부 청 진상 특이사항은 없었고, 복부소견상 간 및 비장은 촉 진되지 않았으며 사지의 부종도 없었다.
검사실 소견: 개인 병원에서 전원되어 처음 내과 입 원 당시 실시한 혈액 검사상 혈색소 6.3g/dL, 헤마토크 리트 19%, MCV, MCH, MCHC는 각각 101.0fL, 34.0pg, 34.0g/dL이었고, 망상적혈구는 0.3%로 감소되어 있었 다. 백혈구는 4,610/μL (호중구 52%, 림프구 38%, 단 구 8%, 호산구 2%), 혈소판은 377,000/μL으로 정상이 었으며, 말초혈액도말 소견상 경도의 이형적혈구증가 증과 부동적혈구증가증을 보이는 대구성 정색소성 빈 혈상이 관찰되었다. 생화학 검사에서 총단백 8.1g/dL, 알부민 4.1g/dL, 총빌리루빈 0.3mg/dL, alkaline phos- phatase 73U/L, AST 35U/L, ALT 22U/L, LDH 179U/L, 철 116μg/dL, 총철결합능 101μg/dL, ferritin 167.5ng/
mL이었다. 갑상선 기능 검사상 총 T3는 166.85ng/dL 로 정상이었으나, 총 T4와 유리 T4는 각각 1.49μg/dL, 0.18ng/dL로 감소되어 있었으며, TSH는 72.91mIU/L로 증가되어 있었다. 항갑상선글로불린 항체와 항갑상선 과산화효소 항체(anti-thyroperoxidase antibody)는 각 각 2,000.0 이상 U/mL, 1,000.0 이상 U/mL로 높게 측정 되었다.
하시모토병으로 인한 갑상선기능저하증 및 동반된 빈혈로 진단 받고 1차 입원 5일째 levothyroxine 25μg
부터 시작하여 입원 9일째 levothyroxine 100μg로 증 량 후 퇴원하였다. 1차 입원 당시 levothyroxine투여 외 에 수혈을 포함한 다른 특별한 치료는 하지 않았다.
Levothyroxine 100μg 복용하면서 외래 경과 관찰 중 levothyroxine 투여 시작 후 5주째 시행한 혈액검사상 빈혈이 악화된 소견을 보여 재입원하였다.
재입원 후 시행한 혈액 검사상 혈색소 3.5g/dL, 헤마 토크리트 11%, MCV 118.0fL, MCH 39.0pg, MCHC 33.0g/dL, 망상적혈구는 0.8%, 백혈구 4,630/μL (호중 구 64%, 림프구 35%, 단구 1%), 혈소판 241,000/μL이 었다. 말초혈액도말 소견상 1차 입원 시와 마찬가지로 경도의 이형적혈구증가증과 부동적혈구증가증을 보이 는 대구성 정색소성 빈혈상이 관찰되었다. 생화학 검 사상 총단백 7.6g/dL, 알부민 3.9g/dL, 총빌리루빈 0.8mg/
dL, alkaline phosphatase 66U/L, AST 18U/L, ALT 11U/L, LDH 199U/L, 철 123μg/dL, 총철결합능 127μg/
dL, ferritin 273.8ng/mL이었고, 비타민 B12 및 엽산 농 도는 각각 552.0pg/mL, 19.3ng/mL로 정상이었다.
HBsAg, HBsAb, HCV Ab 및 항핵항체, human im- munodeficiency virus (HIV) Ab, parvovirus PCR 등은 모두 음성이었으며, 류마티스인자는 10IU/mL로 정상 이었고, Epstein-Barr virus (EBV)의 viral capsid antigen (VCA)에 대한 IgM항체는 음성, IgG항체는 양성이었 다. 갑상선 기능 검사상 총 T3 88.69ng/dL, 총 T4 10.17 μg/dL, 유리 T3 2.28pg/mL, 유리 T4 1.65ng/dL, TSH 0.40mIU/L이었다.
골수검사 소견: 골수검사가 빈혈 악화로 재입원 후
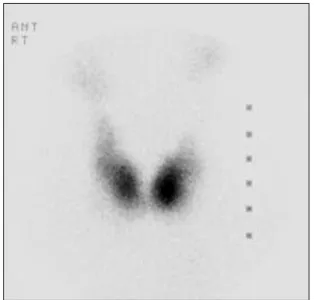
Fig. 2. Radionuclide (99mTc) scan of the thyroid showing diffuse thyroid enlargement with irregular contour.
3일째 시행되어졌는데, 과립구계와 거핵구계는 정상 소견을 보였으나 적아구는 매우 감소되어 M:E비가 9:1, 세포충실도는 40%로서 진성적혈구무형성증에 합당한 소견이었다(Fig. 1).
영상검사 소견: 단순 흉부촬영에선 경도의 경부 기 도 협착 소견 외 특이 소견은 없었으며, 경부 전산화단 층촬영상 불균일한 조영 증강을 보이는 미만성의 갑상 선 종대 및 그로 인한 경도의 기도 압박이 관찰되었고, 다른 종괴나 림프선 비대 소견은 없었다. 갑상선 스캔 소견상 갑상선은 약간 커져 있고 양 엽의 윤곽의 기형 이 심하며 전체 흡수율은 4.2%로 정상 범위를 보였다(Fig.
2).
치료 및 경과: 재입원 1일째(levothyroxine 투여 시작 후 약 5주째) 총 3단위의 적혈구 수혈을 시행 받은 후 혈색소 6.2g/dL, 헤마토크리트 19%였으며, 5일째 2단 위의 수혈을 더 받은 후 혈색소 9.0g/dL, 헤마토크리트 28%로 재입원 8일째 퇴원하였다. 재입원 중 및 퇴원 후에도 계속 levothyroxine 100μg를 매일 복용하였다.
외래 추적 관찰 중 levothyroxine 투여 시작 후 약 8주 째 시행한 혈액 검사상 혈색소 9.8g/dL, 헤마토크리트 31%, MCV 102.0fL, MCH 33.0pg, MCHC 32.0g/dL였으 며, levothyroxine 투여 시작 후 약 13주째에는 혈색소 11.2g/dL, 헤마토크리트 34%, MCV 101.0fL, MCH 33.0pg, MCHC 33.0g/dL, 약 20주째에는 혈색소 12.6g/
dL, 헤마토크리트 38%, MCV 97.0fL, MCH 32.0pg, MCHC 33.0g/dL로 점점 회복되는 경향을 보였다.
Levothyroxine 투여 시작 후 약 15주째 시행한 갑상선
기능 검사상 유리 T4 1.25ng/dL, 유리 T3 2.95pg/mL, TSH 4.29mIU/L로 측정되었다.
고 찰
진성적혈구무형성증은 과립구, 거핵구 계통에 이상 이 없이 적혈구 계통만 선택적으로 억제되는 질환으로 말초혈액에서 정구성 정색소성 또는 대구성 정색소성 빈혈 및 망상적혈구의 감소를 보이며, 골수에서 세포 충실도는 정상 또는 증가하나 적혈구 계통의 심한 감 소를 보인다. 진성적혈구무형성증은 1922년에 Kaznel- son에 의하여 처음으로 기술되었으며 이후 pure red cell anemia, isolated aplastic anemia, erythroblast hypo- plasia, erythrophthisis, erythroblastopenia, red cell aplas- tic anemia, red cell agenesis, aregenerative anemia 등의 여러 동의어로 불려 왔다.5) 진성적혈구무형성증은 크 게 선천성인 Diamond-Blackfan 증후군과 후천성인 경 우로 구분되는데 후천성 진성적혈구무형성증 중 가장 흔히 동반되는 질환은 흉선종으로 진성적혈구무형성 증 환자의 약 10∼50%이며 그 외 만성림프구성백혈병 이나 악성림프종과 같은 혈액 종양, 비혈액 종양, par- vovirus B19, HIV, EBV, cytomegalovirus (CMV), mumps, hepatitis virus 등과 같은 감염, 약물이나 화학 물질, 전신홍반루프스, 류마티스관절염 등의 교원성 질환, 임신, 용혈성 빈혈, 신부전증, 심한 영양결핍 등 의 다양한 질환에 의해 생길 수 있다.1,5)
후천성 진성적혈구무형성증의 원인으로 자가면역이 관여한다고 제안되었는데, 이는 적혈구 생성 과정에 영향을 미치는 면역글로불린 억제인자가 혈청에 존재 하며, 면역억제 치료를 함으로써 관해 후 이들이 혈청 에서 사라진다는 사실에 기초를 두고 있다. 또한 진성 적혈구무형성증을 가진 환자의 혈액에서 T 세포의 클 론성 확장이 관찰되고, 이러한 림프구가 적혈구계 집 락 형성을 억제하는 실험실 소견 등은 항체에 의한 기 전 외에 NK 세포(natural killer cell)나 T 세포 매개성 면역학적 기전을 시사한다.6,7) 게다가, 진성적혈구무형 성증이 흉선종, 전신홍반루푸스, 류마티스관절염,2) 복 합내분비샘결핍증후군(multiple endocrine gland in- sufficiency)8) 등을 포함한 다른 면역학적 질환과 자주 동반된다는 사실은 진성적혈구무형성증에 대한 이런 면역학적 기반을 또한 뒷받침한다.
진성적혈구무형성증을 동반할 수 있는 자가면역 질 환으로서 앞서 기술한 질환 외에도 혼합결합조직병,3) 쇼그렌증후군4) 등도 보고되었으나, 저자가 국내 및 영
문 문헌을 고찰한 바로는 본 증례에서와 같이 다른 질 환 없이 하시모토병에 의한 원발성 갑상선기능저하증 과 병발된 진성적혈구무형성증에 대해서는 보고된 바 가 없었다.
영문 문헌 고찰상 진성적혈구무형성증과 원발성 자 가면역성 갑상선기능저하증과의 관련성에 대해 보고 한 다른 증례들에서는, 갑상선기능저하증 외에도 모두 전신홍반루푸스가 동반되어 있었으며, 부신피질호르 몬과 levothyroxine을 병용 치료함으로써 진성적혈구 무형성증이 회복되었다.9-12) 저자들의 예는 항핵항체 가 음성이었으며, 또한 전신홍반루푸스를 의심할 만한 다른 증상 및 징후를 발견할 수 없어 상기 보고들과는 차이를 보이며, 유리 T3와 유리 T4의 감소, TSH 증가, 고농도의 항갑상선글로불린 항체 및 항갑상선과산화 효소 항체, 갑상선 종대 등의 소견으로 하시모토병으 로 인한 원발성 갑상선기능저하증에 동반된 진성적혈 구무형성증으로 진단하였다. 또한 이 환자에서는 면역 억제제 등의 다른 약제의 투여 없이 갑상선호르몬의 지속적인 단독 투여만으로 빈혈의 회복 소견을 보였 다.
체외 실험에서 갑상선호르몬은 적혈구생성인자(ery- thropoietin)가 적혈구계 집락의 형성에 미치는 효과를 촉진하며, 유도된 저산소증으로 인해 적혈구생성인자 의 생성을 증가시킨다. 또한 갑상선 절제술을 시행 받 은 동물에서 빈혈은 골수에서의 적혈구계 저형성과 관 련되어 있었다. 많은 갑상선기능저하증 환자들은 경도 에서 중등도의 빈혈을 보이는데, 이는 갑상선기능저하 에 따른 대사율의 감소, 산소 요구량의 감소, 적혈구생 성인자의 감소에 따른 일종의 적응성 “생리적 빈혈”로 설명하며 종종 골수 세포충실도의 감소와 적혈구계 전 구세포의 감소를 동반한다고 알려져 있으며, 이런 경 우 혈색소 농도가 8∼9g/dl 이하인 경우는 거의 없다.13) 이 환자에서 하시모토병과 진성적혈구무형성증이 동반된 기전에 관해서는 정확히 알 수 없으나, 다른 자 가면역질환에 동반된 진성적혈구무형성증에서처럼 면 역감시체계의 결함으로 인해 어떤 표적 기관, 예를 들 면 적혈구 전구세포나 적혈구생성인자 등에 대한 항체 의 과도한 생성 또는 세포 매개성 면역학적 기전 등이 관여되어 있을 것으로 추정되나 이들에 대한 실험실적 측정은 하지 않았으며, 전술한 바와 같이 갑상선기능 저하증 자체로 인한 생리적인 산소 요구량 감소 및 갑 상선 호르몬 결핍으로 인한 직접적인 적혈구계 저형성 등도 빈혈의 악화에 영향을 주었을 것으로 생각한다.
진성적혈구무형성증의 치료는 이차적인 경우는 가
능한 모든 약물투여를 중지하고, 증상을 야기하지 않 는 범위로 혈색소치를 유지하며, 동반된 질환을 치료 하면서 자연 회복을 기다리는 것으로 되어있다. 흉선 의 비대 또는 흉선종이 있는 경우는 그 진단 및 골수의 회복 등을 목적으로 수술이 권유되며, parvovirus B19 감염에 의한 경우 면역글로불린 투여시 대부분의 환자 에서 효과를 보인다.1,5) 진성적혈구무형성증의 원인을 알 수 없는 경우는 부신피질호르몬, cyclosporine, cy- clophosphamide, azathioprine, antithymocyte globulin (ATG) 등의 순차적인 단독요법 또는 병용요법이 사용 되며, 이외에도 비장적출술, 안드로겐, 혈장분리교환 술(plasmapheresis) 등이 시도되기도 한다.5) 또한 최근 에는 다른 약제들에 불응성인 경우 rituximab (anti- CD20 monoclonal antibody), alemtuzumab (anti-CD52 monoclonal antibody), fludarabine, cladribine 등에 대한 효과도 보고되고 있다.5,14,15)
특이할 만한 사실은 이 환자에서는 다른 자가면역질 환에 동반된 진성적혈구무형성증 시 흔히 사용하는 부 신피질호르몬 등의 면역억제치료 없이 갑상선호르몬 제제의 단독 투여만으로 적혈구 조혈 기능이 활성화되 었다는 것인데, 투여된 갑상선호르몬이 다른 면역체계 와 어떤 기전에 의해 진성적혈구무형성증의 회복에 기 여하였는지에 대해서는 추후 더 연구가 필요할 것으로 생각한다.
요 약
후천성 진성적혈구무형성증(acquired pure red cell aplasia)은 골수에서 백혈구계 및 거핵구계의 성숙이나 수에는 이상이 없이 적혈구계의 선택적인 심한 결핍을 나타내는 드문 혈액 질환이다. 이 질환의 원인으로 자 가면역이 관여한다고 제시되고 있으며, 진성적혈구무 형성증이 전신홍반루푸스, 류마티스관절염, 흉선종, 혼합결합조직병, 쇼그렌증후군 등 다양한 면역질환과 연관되어 발생한다는 사실은 이를 뒷받침한다. 그러 나, 하시모토병(Hashimoto disease)과 진성적혈구무형 성증이 다른 동반된 자가면역질환 없이 병발된 예는 국내 및 영문 문헌 고찰상 보고된 바가 없다. 이에 저 자들은 62세된 여자 환자에서 하시모토병으로 인한 갑상선기능저하증과 동반된 진성적혈구무형성증을 진 단하고, 갑상선호르몬제제의 단독 투여만으로 조혈기 능의 회복을 보인 증례를 경험하였기에 문헌 고찰과 함께 보고하는 바이다.
참 고 문 헌
1) Fisch P, Handgretinger R, Schaefer HE. Pure red cell aplasia. Br J Haematol 2000;111:1010-22.
2) Rodrigues JF, Harth M, Barr RM. Pure red cell aplasia in rheumatoid arthritis. J Rheumatol 1988;
15:1159-61.
3) Julkunen H, Jantti J, Pettersson T. Pure red cell aplasia in mixed connective tissue disease. J Rheu- matol 1989;16:1385-6.
4) Ibkhatra S, Jacobsson L, Manthorpe R. The associa- tion of pure red cell aplasia and primary Sjogren’s syndrome. Clin Exp Rheumatol 1997;15:119-20.
5) Dessypris EN. Pure red cell aplasia. In: Hoffman R, Benz EJ, Shattil SJ, eds. Hematology: basic princi- ples and practice. 4th ed. Philadelphia, USA: Else- vier Inc., 2005:429-39.
6) Yamada O. Clonal T cell proliferation in patients with pure red cell aplasia. Leuk Lymphoma 1999;
35:69-82.
7) Handgretinger R, Geiselhart A, Moris A, et al. Pure red-cell aplasia associated with clonal expansion of granular lymphocytes expressing killer-cell inhibi- tory receptors. N Engl J Med 1999;340:278-84.
8) Myers TJ, Bower BF, Hild DH. Pure red cell aplasia and the syndrome of multiple endocrine gland in-
sufficiency. Am J Med Sci 1980;280:29-34.
9) Francis DA. Pure red-cell aplasia: association with systemic lupus erythematosus and primary auto- immune hypothyroidism. Br Med J (Clin Res Ed) 1982;284:85.
10) Heck LW, Alarcon GS, Ball GV, et al. Pure red cell aplasia and protein-losing enteropathy in a patient with systemic lupus erythematosus. Arthritis Rheum 1985;28:1059-61.
11) Franzen P, Friman C, Pettersson T, Fyhrquist F, Ruutu T. Combined pure red cell aplasia and pri- mary autoimmune hypothyroidism in systemic lupus erythematosus. Arthritis Rheum 1987;30:839-40.
12) Rahman J, Rashid MA, Yunus AB, et al. Acquired pure red cell aplasia--a case report. Bangladesh Med Res Counc Bull 1998;24:79-81.
13) Gregg XT, Prchal JT. Anemia of endocrine disor- ders. In: Lichtman MA, Kipps TJ, Kaushansky K, Beutler E, Seligsohn U, Prchal JT, eds. Williams hematology. 7th ed. New York, USA: McGraw-Hill Co., 2006:459-60.
14) Ahn J, Lee K, Lee J, et al. A case of refractory idio- pathic pure red cell aplasia responsive to fludarabine treatment. Br J Haematol 2001;112:527-9.
15) Robak T, Kasznicki M, Blonski JZ, Dmoszynska A, Skotnicki AB. Pure red cell aplasia in patients with chronic lymphocytic leukaemia treated with cladri- bine. Br J Haematol 2001;112:1083-5.