Printed in the Republic of Korea
http://dx.doi.org/10.5012/jkcs.2013.57.6.703
Selection of Suitable Micellar Catalyst for 1,10-Phenanthroline Promoted Chromic Acid Oxidation of Formic Acid in Aqueous Media at Room Temperature
Aniruddha Ghosh, Rumpa Saha, Sumanta K. Ghosh, Kakali Mukherjee, and Bidyut Saha*
Homogeneous Catalysis Laboratory, Department of Chemistry, The University of Burdwan, Golapbag, Burdwan, Pin 713104, WB, India. *E-mail: [email protected]
(Received October 8, 2012; Accepted October 28, 2013)
ABSTRACT. In the present investigation, kinetic studies of oxidation of formic acid with and without catalyst and promoter in aqueous acid media were studied under the pseudo-first order conditions [formic acid]T >>[Cr(VI)]T at room temperature.
In the 1,10-phenanthroline (phen) promoted path, the cationic Cr(VI) phen complex is the main active oxidant species under- goes a nucleophilic attack by the substrate to form a ternary complex which subsequently experiences a redox decomposition through several steps leading to the products CO2 and H2 along with the Cr(III) phen complex. The anionic surfactant (i.e., sodium dodecyl sulfate, SDS) and neutral surfactant (i.e., Triton X-100, TX-100) act as catalyst and the reaction undergo simultaneously in both aqueous and micellar phase with an enhanced rate of oxidation in the micellar phase. Whereas the cat- ionic surfactant (i.e., N-cetyl pyridinium chloride, CPC) acts as an inhibitor restricts the reaction to aqueous phase. The observed net enhancement of rate effects has been explained by considering the hydrophobic and electrostatic interaction between the sur- factants and reactants. The neutral surfactant TX-100 has been observed as the suitable micellar catalyst for the phen pro- moted chromic acid oxidation of formic acid.
Key words: Formic acid, [Cr(VI)]T, Cr(VI)–phen, TX-100, SDS
Organic and inorganic salts of Cr(VI) are well-known oxidants for organic compounds.1 However, these salts are rather drastic and non-selective oxidants. Further, they are insoluble in most organic solvents. Thus miscibility is a problem.2 Selective oxidation of organic compounds is an important reaction in synthetic organic chemistry.3 Chro- mium chemistry is mainly dominated by two oxidation states: Cr(III) and Cr(VI). In addition to non-toxic trivalent chromium some amount of hexavalent chromium remains in the industrial effluents.4 There are two problems. Hexava- lent chromium is carcinogenic5,6 and nonaqueous medium is not environmental friendly.7 Use of large excess of sub- strate over hexavalent chromium and aqueous medium can solve the problem. Sometimes various chelating agents are often used as promoter (Picolinic acid, 2,2'-bipyridine, 1,10- phenanthroline) for enhancement the transformation pro- cess.8−16 A fundamental concern in any chemical process is the yield of a particular transformation - the effectiveness of converting given substrates into a single desired product.
Therefore, control of selectivity to the desired product would be highly advantageous.17 The choice of K2Cr2O7 in aque- ous micellar media is due to the control and selective oxi- dation of formic acid by maintaining pseudo-first order criteria. The catalysis of reaction by organized media is of increasing interest.18−21 The search for improved processes
generating hydrogen from liquid fuels at higher reaction rates and under milder, well-controlled conditions is an active area of interest in the fuel cell field. Among possible liquid fuel candidates, formic acid (HCOOH) definitely shows a great potential as in situ source of hydrogen for fuel cells.22 Treatment of formic acid with sulfuric acid is a convenient laboratory source of CO. In general, formic acid can be decomposed via dehydrogenation23 (HCOOH→H2+ CO2).
Recently, the decomposition of formic acid has been intensely investigated for hydrogen generation, particularly in the area of reforming catalyst development.24 For example, metal complex-based homogeneous catalysts have been reported to have high performance for the formic acid decomposition at near ambient temperatures.25−28 Homo- geneous catalysts are easy to use widely because of ease to be controlled, retrieved, and recycled and there is a strong desire to develop homogeneous catalysts with high activity and selectivity for the formic acid decomposition by taking water as eco-friendly solvent under mild conditions. It is generally accepted that formic acid is oxidized to CO2 via the so-called dual-pathway mechanism: a direct path to CO2 via a reactive intermediate and an indirect path via a poisoning species which is oxidized to CO2. On the other hand, the reactive intermediate in the direct path has not been defined yet.29
For catalytic process micellar solution provides a way for alternative synthetic routes in an aqueous medium. In an aqueous phase surfactant molecules aggregate at ambient conditions, forming micelles with a hydrophobic core and hydrophilic corona.30 A micellar system appears to be homogeneous since these aggregates are of colloidal size;
however in reality the absorbed reactants are in a micro het- erogeneous two phase system. The catalytic efficiency will be governed by the affinity of the reactivity of the bound reagent molecules.13 The molecule is composed of a non- polar hydrophobic portion and a polar hydrophilic portion.
In solution mutual interactions between surfactant mole- cules, solvent molecules and surfactant and solvent mole- cules decide the ultimate state. When self-interaction of both surfactant and solvent molecules cannot be compensated by their mutual interactions, the surfactant molecules tend to associate in a regular pattern forming “association colloids”
or “micelles”. The surfactants that form micelle can be basi- cally classified as (i) Cationic (ii) Anionic and (iii) Non- ionic. All the three types of surfactants are used as micellar catalyst in this present work. Micellar catalysis has also received considerable attention in view of analogies drawn between micellar and enzyme catalysis21. As the title reaction is very slow, suitable combination of micellar catalyst13,31−33 and promoter15,16,21,34−36 are used to speed up the reaction.
The micellar catalyzed PA and bipy promoted formic oxi- dation by chromic acid was studied previously.14,37
In the present study, we mainly focus our attention on the selection of the suitable micellar catalyst one among the three surfactant N-cetylpyridinium chloride (CPC), sodium dodecyl sulfate (SDS) and Triton X-100 (TX-100) in com- bination with 1,10-phenanthroline (phen) as promoter in aqueous acidic media.
Under the kinetic and experimental conditions, carbon dioxide was qualitatively detected by purging dinitrogen14,37,38 through the reaction vessel and passing the out coming gas through a narrow tube containing Ca(OH)2. Under the experimental conditions, polymerization of acrylonitrile was indicated under a nitrogen atmosphere. Hence, posi- tive result for acrylonitrile polymerization indicates free radical formation.14,37
The overall stoichiometry of the reaction may be repre- sented as:
3HCOOH + 2HCrO4−+ 8H+→ 2Cr(III) + 3CO2+ 8H2O Both in the presence and absence of phen, under the experimental conditions, [formic acid]T >> [Cr(VI)]T, (subscript T stands for the total concentration), the rate of decay of Cr(VI) shows a first order dependence on
[Cr(VI)]. This first order dependence on Cr(VI) is also maintained in the presence of surfactants CPC, SDS and TX-100. The pseudo- first-order rate constants (kobs) have been evaluated from the linear plot of log[Cr(VI)]T versus time (t) as usual.
Dependence of substrate and acid on the observed rate both in presence and absence of promoter and catalyst were already established14,37. The rate is first-order with respect to formic acid. Product analysis and spectroscopic findings helps us to indicate the mechanism of the formic acid oxi- dation. The mechanism of the reaction can be divided into two parts: (i) unpromoted path and (ii) promoted path. The unpromoted path for chromic acid oxidation of formic acid (Scheme 1) was established earlier.14,37
The gradual production of the Cr(III) and disappearance of Cr(VI) species were confirmed spectrophotometrically.
The colors of the final solutions in the absence of phen and in the presence of the phen are distinctly different indicat- ing different types of species. In the unpromoted path the final color observed as pale blue (λmax = 582 nm and λmax = 408 nm; Fig. 1) and the corresponding transitions8,39 are 582 nm for 4A2g(F)→4T2g(F) and 408 nm for 4A2g(F)→4T1g(F) of Cr(III) species. Whereas the color observed in phen pro- moted path was pale violet (λmax = 550 nm, Fig. 1) due to the transition 4A2g(F)→4T2g(F) of Cr(III) species. The spec- tra of the final solution in absence of promoter and pure chromic sulfate solution in aqueous sulfuric acid media are almost similar, but it is different due to the presence of dif- ferent types of Cr(III) species (λmax = 550 nm for phen pro- moted path Fig. 1). This simply indicates that the final
Figure 1. Absorption spectrum of reaction mixture (after com- pletion of reaction): [formic acid]T= 75×10−4 mol dm−3, [Cr(VI)]T = 5×10−4 mol dm−3, [H2SO4]T = 0.5 mol dm−3, Temp = 30oC (i) Unpromoted: (The spectrum of the chromic sulfate is identical with this under the experimental condition), (ii) phen promoted:
[phen]T = 0.01 mol dm−3.
Cr(III)-aqua species is Cr(III)−phen complex. In the phen promoted reaction path, there is a hypsochromic (blue) shift for the transition 4A2g(F)→4T2g (F) compared to the final solution of the unpromoted reaction path (Fig. 1). The blue
shift in the Cr(III)−phen complex for 4A2g(F)→4T2g(F) transition is due to the presence of the strong field ligand phen compared to ligand water. In the Cr(III)−phen com- plex,34 the band due to the transition 4A2g (F)→4T1g(F)
Figure 2. (a) Scanned absorption spectra of the reaction mixture in absence of promoter (phen) at regular time intervals (10 min). [for- mic acid]T = 75×10−4 mol dm−3, [Cr(VI)]T = 5×10−4 mol dm−3, [H2SO4]T = 0.5 mol dm−3. Temp = 30oC. (b) Scanned absorption spectra of the reaction mixture in absence of promoter (phen) at regular time intervals (5 min). [formic acid]T = 75×10−4 mol dm−3, [Cr(VI)]T
= 5×10−4 mol dm−3, [H2SO4]T = 0.5 mol dm−3. [SDS] = 0.04 mol dm−3, Temp = 30oC. (c) Scanned absorption spectra of the reaction mixture in absence of promoter (phen) at regular time intervals (5 min). [formic acid]T = 75×10−4 mol dm−3, [Cr(VI)]T = 5×10−4 mol dm−3, [H2SO4]T = 0.5 mol dm−3. [TX-100] = 0.02 mol dm−3, Temp = 30oC. (d) Scanned absorption spectra of the reaction mixture in presence of promoter (phen) at regular time intervals (1 min). [formic acid]T = 75×10−4 mol dm−3, [Cr(VI)]T = 5×10−4 mol dm−3, [H2SO4]T
= 0.5 mol dm−3. [phen]T = 0.01 mol dm−3. [SDS] = 0.04 mol dm−3, λisosbestic = 523 nm, Temp = 30oC. (e) Scanned absorption spectra of the reaction mixture in presence of promoter (phen) at regular time intervals (3 min). [formic acid]T = 75×10−4 mol dm−3, [Cr(VI)]T
= 5×10−4 mol dm−3, [H2SO4]T = 0.5 mol dm−3. [phen]T = 0.01 mol dm−3. [TX-100] = 0.02 mol dm−3, λisosbestic = 517 nm, Temp = 30oC.
merges with the charge-transfer band (Fig. 1). The appear- ance of the charge-transfer band at much lower energy (visible range) in the observed UV-vis spectrum for the proposed Cr(III)−phen complex is consistent with the favoured metal to ligand charge-transfer. The vacant π*
(antibonding) M.O. of the ligand (phen) strongly pro- motes the metal to ligand40−42 electron-transfer (MLCT).
The formation of the charge-transfer band (MLCT) at this lower energy (higher wavelength) for the phen promoted path supports the existence of the Cr(III)−phen complex in the final solution.
The reaction mixture were scanned in the range 400−700 nm in both presence and absence of phen and surfactant at reg- ular time intervals (10 minute) to follow the gradual devel- opment of the reaction intermediates (if any) and the product.
The scanned spectra (Fig. 2a) indicates the gradual disap- pearance of Cr(VI) species and appearance of Cr(III) spe- cies in the unpromoted path. The scanned spectra were also recorded for the presence of SDS and TX-100 sep- arately both in presence and absence of phen (Fig. 2b, 2c, 2d and 2e).
It is interesting to note that under the present experimen- tal and kinetic conditions, appearance of the single isosbestic point in each scanned spectra nicely suggests the formation of short-lived intermediates like Cr(V) and Cr(IV) in very low concentration43 during the oxidation. In such cases, because of the very low concentration of the species, it may not always be possible to isolate or detect the species spec- trophotometrically.44 In the phen-promoted reaction path
the active oxidant involved in the oxidation process is Cr(VI)−phen which supports the phen promoted reaction mechanism15,16,21 (Scheme 2, Fig. 3). The presence of the charge-transfer band (MLCT) at the lower energy (higher wavelength) for the Cr(VI) spectra in presence of phen compared to spectra of only Cr(VI) and only phen supports the formation of the active oxidant Cr(VI)−phen complex during the reaction. The absorbance spectra of the solution without substrate (formic acid) is found due to phen only is in between the spectra of Cr(VI) in absence of phen and Cr(VI) in presence of phen.
In the unpromoted path the observed rate law14,37 for Scheme 1 is given by:
kobs(u) = (2/3) k1K1K2 [formic acid]T[H+]2
In the proposed cyclic transition state, the reduction of Cr(VI) to Cr(IV) occurs through hydride or hydrogen ion transfer. In the next step, Cr(IV) is further reduced to Cr(III) by different possible routes.14,37
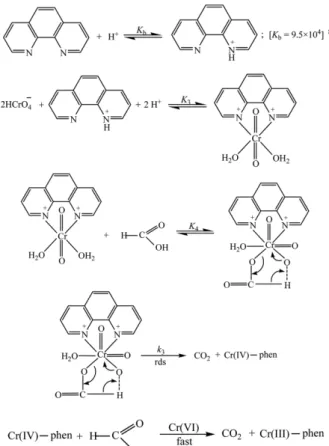
In the phen promoted reaction path the formation of the Cr(III)−phen complex detected spectrophotometrically indi- cates that the promoter (phen) used here undergo complex- ation with the highest oxidation states of chromium i.e.
Cr(VI) (labile, t2g0 eg0). Based on this concept, it is believed that the Cr(VI)−phen complex produced in the pre-equilib- rium step is the active oxidant (mentioned as AO+ in Scheme 2). The Cr(VI)−phen complex reacts with formic acid to form a ternary complex and would undergo redox decomposition via several steps to produce Cr(III)−phen complex. The mechanism of the phen promoted reaction path can be schematized as follows:
Figure 3. Absorption spectrum of reaction mixture with and without promoter (in absence of substrate): (i) [Cr(VI)]T = 5×10−4 mol dm−3, [H2SO4]T = 0.5 mol dm−3. (ii) [phen]T = 0.01mol dm−3. (iii) [Cr(VI)]T = 5×10−4 mol dm−3, [H2SO4]T = 0.5 mol dm−3, [phen]T
= 0.01mol dm−3. Temp = 30oC.
Scheme 1. Cr(VI) oxidation of formic acid in absence of phen.
The rate law of phen promoted reaction path can be pro- posed as:
kobs(phen) = (2/3) k3K3K4 [formic acid]T[phen]T[H+]
Surfactants are the amphiphilic molecules which undergo special type of self-assembly process, and the phenome-
non is known as micellization.45 Model composed of amphiphilic materials and aggregated colloids provide a useful way of better understanding in many invaluable areas such as biochemistry, medicine, and pharmaceuticals as well as in catalysis.46,47 Several research groups48,49 have studied the kinetics of organic reactions in the presence of surfactant micelles as these can influence the rate of the chemical reaction incorporating the reactant molecules in micellar pseudo-phase.50
The partitioning of the neutral ester [(1) Scheme 1] in all types of the surfactants is more or less equally probable (Fig. 4-1) in unpromoted reaction path.
The fundamental process in micellar catalysis or inhibi- tion is the counter ion binding with micelle. Micelles can either attract the reactive species or repel them depending upon the electrical charge distribution on their head groups.
Thus, micelles may bring the solubilized substrate and reac- tive ions together or keep apart such that the reactions are catalyzed or inhibited. Micellar catalysis of reactions in aqueous solutions is usually explained on the basis of a dis- tribution of reactants between water and the micellar pseudo- phase. The reaction occurs at different rates in the two media.
If the solubility of the reactants is greater in the micelles than in water then the local concentration is increased in the micelle, often with suitable orientation of the reactants bound in the micelle. Further, it was thought that only elec- trostatic interaction occurs between micelles and reactants, however, later it was revealed that reaction rates are also affected by hydrophilic interactions. Surfactant inhibition dependence of rate constant on [CTAB] we propose a model8,16 for bimolecular reactions. Partitioning of proton in unpromoted reaction path is highest for anionic surfac- Scheme 2. Cr(VI) oxidation of formic acid in presence of phen.
Figure 4-1. Schematic representation of partitioning of neutral ester and proton in (a) Cationic surfactant, (b) Anionic surfactant and (c) Neutral surfactant.
tant SDS due to favourable and very strong electrostatic attraction. Thus for SDS, the reaction can go both in aque- ous and micellar phase where the active reactants are pref- erably accumulated.10,13 Thus, SDS allows the reaction to proceed in both aqueous and micellar interphases.16,51 Small H+ can easily penetrate in SDS micelle due to the effective electrostatic attraction. On the other hand a severe electrostatic repulsion between the approaching H+ ion and the positive hydrophilic head group of cationic CPC makes unavailable for the effective concentration of neutral ester and H+ within the micellar core. Consequently, the reaction is restricted only in aqueous phase where concentration of the ester species is depleted due to its preferential parti- tioning in the micellar pseudo-phase. Thus CPC shows the rate retarding effect even from pure water.44 TX-100, a neu- tral surfactant catalyzes the rate of oxidation process slightly higher than pure water, as there are no charge head groups are present.16
The most pronounced acceleration for the formic acid oxidation process is observed in the TX-100 catalyzed phen promoted path (Table 1).
The Cr(VI)−phen complex represented as AO+ (Fig. 4-2, Scheme 2), a cationic complex, has been argued as the active oxidant in the promoted path. Because of the elec- trostatic attraction it can preferably be distributed in the micellar pseudo-phase of the anionic surfactant SDS. Thus, in the presence of SDS micelle, the oxidation process can propagate in both the micellar pseudo-phase (where both the active oxidant and substrate are preferably concentrated) and aqueous phase to give the observed rate acceleration.14,37,44 In this work the highest acceleration of rate is observed for combination of TX-100 micelles and phen. This is explained on the basis of (Fig. 4-2) the degree of formic acid binding to the nonionic surfactant micelles is probably higher than the degree of binding to the anionic ones.13 The hydrophobic part of TX-100 micelles are larger compared to SDS, so maximum number of Cr(VI)−phen (active oxidant) com- plex can accumulate into the TX-100 micellar core. Here the active oxidant Cr(VI)−phen complex react with formic acid to form a ternary complex which undergoes redox decomposition in a rate-limiting step52,53 giving rise to the product. The highest degree of rate acceleration in TX-100 Table 1. kobs and half life of the reaction in presence and absence of bipy and non-functional micellar catalyst
Promoter [mol dm−3] Micellar catalyst [mol dm−3] 104×kobs (sec−1) t1/2 (h)
None None 0.02875 ± 0.003 66.7
1,10-phenanthroline (phen) 0.01 None 0.159 ± 0.003 12.1
None SDS 0.04 0.2266 ± 0.006 8.49
None TX-100 0.02 0.1816 ± 0.005 10.6
None CPC 0.002 0.01123± 0.005 171.41
1,10-phenanthroline (phen) 0.01 SDS 0.04 6.445 ± 0.004 0.29
1,10-phenanthroline (phen) 0.01 TX-100 0.02 17.185 ± 0.003 0.11
[Cr(VI)]T = 5×10−4 mol dm−3, [H2SO4]T = 0.5 mol dm−3, [formic acid]T = 75×10−4 mol dm−3, Temp = 30oC.
Figure 4-2. Schematic representation of partitioning of substrate and active oxidant [AO+ = Cr(VI)-promoter complex] in (a) Cationic surfactant, (b) Anionic surfactant and (c) Neutral surfactant.
catalyzed phen promoted path is most probably due to the maximum accumulation of Cr(VI)−phen complexes which forcefully allows to react with formic acid within the large hydrophobic core.16,54 In this study combination of CPC and phen are not used due to rate retardation effect of CPC in absence of promoter.
The pseudo-first-order rate constants (kobs, s−1) were deter- mined from the slope of plots of ln(A450) against time (t) at wavelength 450 nm. The t1/2 values are directly calculated in Table 1 by using relation t1/2= (ln2/kobs), where ln2 = 0.693, kobs = pseudo-first-order rate constant.
From the Table 1 it is observed that the normal reaction is very much sluggish whereas the rate is increased upon introduction of 1, 10-phenanthroline in the reaction sys- tem in the presence and absence of micelles. Rate enhancement is small for TX-100 compared to SDS in absence of phen. But in presence of phen the rate is max- imum resulting t1/2 is minimum as reflected from the Table 1. On the other hand CPC retards the oxidation process as it is evident from the t1/2 value in the Table 1. When com- bination of SDS (at a fixed concentration; 0.04 mol dm−3) and phen are used together, the total time taken to com- plete the oxidation process is lowered compared to the uncatalyzed and unpromoted reaction. Now similar situ- ation arises when the reaction is catalyzed by TX-100 (a neutral surfactant) instead of SDS, the rate is maximum increased when combination of TX-100 and phen are used. The reaction is accelerated through such an extent that the t1/2 is minimum and it is least (0.11 h or 6.6 min) when the concentrations of TX-100 and phen are 2×10−2 mol dm−3 and 100×10−4 mol dm−3 respectively.
EXPERIMENTAL
1,10-Phenanthroline (AR, Merck, Mumbai, India), formic acid (AR, Qualigens, Mumbai, India), K2Cr2O7 (AR, BDH, India), H2SO4 (AR, Qualigens, Mumbai, India), sodium dodecyl sulfate (AR, SRL, Mumbai, India), TX-100 (AR, SRL, Mumbai, India) and all other chemicals used were of highest degree of purity available commercially. All the solutions were prepared in double distilled water. Solutions of the oxidant and reaction mixtures containing the known quantities of the substrate (s) (i.e., formic acid), promoter (1,10-phenanthroline) under the kinetic conditions [formic acid] >> [Cr(VI)]T. Acid and other necessary chemicals were separately thermostated (± 0.10oC). The reaction was initiated by the requisite amounts of the oxidant with the reaction mixture. Progress of the reaction was monitored by following the decay of Cr(VI) at 450 nm wavelength at dif-
ferent time intervals with the UV-vis [UV-VIS-NIR-3600 (SHIMADZU)] spectrophotometer equipped with a temper- ature controller. Quartz cuvettes of path length 1 cm were used. The pseudo-first-order rate constants (kobs, s−1) were calculated from the slope of plots of ln(A450) versus time (t) which were linear.11 The scanned spectra and spectrum after completion of the reaction were recorded with a UV-vis spectrophotometer [UV-1800 and UV-VIS-NIR-3600 (SHI- MADZU)]. A large excess (15-fold) of reductant was used in all kinetic runs. No interference was observed due to other species at 450 nm.
Surfactant micelles provide an unusual medium, which may affect the rate of reaction. One of the most important properties of micellar system is their ability to affect the rate of the chemical reaction. The effect of surfactants on reac- tion kinetics is called micellar catalysis involves several contributing factors. Notably, formic acid has been widely used as hydrogen source in liquid-phase transfer hydroge- nation reactions of carbon dioxide in the presence of base such as amines under ambient conditions. The excess for- mic acid can be reoxidized to get the deserved product with further addition of chromic acid, maintaining the same reac- tion condition. Obviously, electrostatic attraction/repulsion plays an important role during the course of the reaction in the presence of ionic surfactants. All the surfactant con- centrations used during each set of experiments are above their critical micelle concentration (CMC). The CMC values of all the three surfactants are known from earlier literature.
So CMC values are very helpful to prepare the solutions of surfactants in particular concentration. Combination of TX- 100 micelle and phen promoter enhances the reaction almost 600 times faster compared to the uncatalyzed and unpro- moted reaction. The mechanistic paths of uncatalyzed unpro- moted and SDS catalyzed phen promoted chromic acid oxidation of glycerol have also been compared. SDS micelle in absence of phen enhances the oxidation rate compared to the TX-100 micelle. But in combined reaction mixture i.e., in presence of phen the reaction rate is highly accelerates in TX-100 micelle compared to SDS micelle. It is also noted that the cationic micelle CPC retards the reaction compared to the uncatatalyzed and unpromoted reaction. The Cr(VI)− phen complex, a cationic species has been found to act as the active oxidant in the phen-promoted chromic acid oxi- dation of formic acid. The generation of the final Cr(III) species after the completion of the reaction has been inves- tigated by a series of spectral observation by UV-visible spectrophotometer. The present method of formic acid oxi- dation is simple, accurate, rapid, economical, and precise.
In conclusion, it can be said that neutral micelle TX-100 is
an efficient micellar catalyst for the phen promoted chro- mic acid oxidation of formic acid.
Acknowledgments. Thanks to UGC, New Delhi and CSIR, New Delhi for providing financial help in the form of project and fellowship. Thanks also to The University of Burdwan, Burdwan, India for providing infra-structural facil- ities and the publication cost of this paper was supported by the Korean Chemical Society.
REFERENCES
1. Sundaram, S.; Raghavan, P. S. Chromium-VI reagents:
Synthetic Application; Springer, New York, 2011.
2. Purohit, P.; Kumbhani, S.; Shashtri, I.; Banerjee, K. K.;
Sharma, P. K. Indian J. Chem. 2008, 47, 1671.
3. Banerji, J.; Kotai, L.; Banerji, K. K. Indian J. Chem.
2009, 48, 797.
4. Saha, B.; Orvig, C. Coord. Chem. Rev. 2010, 254, 2959.
5. Holmes, A. L.; Wise, S. S.; Wise, J. P. Indian J. Med. Res.
2008, 128, 353.
6. Saha, R.; Nandi, R.; Saha, B. J. Coord. Chem. 2011, 64, 1782.
7. Das, A. K. Coord. Chem. Rev. 2004, 248, 81.
8. Ghosh, A.; Saha, R.; Mukhejee, K.; Ghosh, S. K.; Bhat- tacharyya, S. S.; Laskar, S.; Saha, B. Int. J. Chem. Kinet.
2013, 45, 175.
9. Basu, A.; Ghosh, S. K.; Saha, R.; Ghosh, A.; Ghosh, T.;
Mukherjee, K.; Bhattacharyya, S. S.; Saha, B. Tenside Surf.
Det. 2012, 49, 481.
10. Ghosh, S. K.; Saha, R.; Ghosh, A.; Mukherjee, K.; Saha, B. Tenside Surf. Det. 2012, 49, 370.
11. Ghosh, S. K.; Ghosh, A.; Saha, R.; Saha, B. Tenside Surf.
Det. 2012, 49, 296.
12. Saha, R.; Ghosh, S. K.; Ghosh, A.; Saha, I.; Mukherjee, K.; Basu, A.; Saha, B. Res. Chem. Intermed. 2013, 39, 631.
13. Ghosh, S. K.; Saha, R.; Mukherjee, K.; Ghosh, A.; Bhat- tacharyya, S. S.; Saha, B. J. Korean Chem. Soc. 2012, 56, 164.
14. Ghosh, S. K.; Basu, A.; Saha, R.; Nandi, R.; Saha, B.
Current Inorg. Chem. 2012, 2, 86.
15. Ghosh, S. K.; Basu, A.; Saha, R.; Ghosh, A.; Mukherjee, K.; Saha, B. J. Coord. Chem. 2012, 65, 1158.
16. Ghosh, A.; Saha, R.; Mukhejee, K.; Ghosh, S. K.; Bhat- tacharyya, S. S.; Saha, B. J. Chem. Res. 2012, 36, 347.
17. Dimitratos, N.; Villa, A.; Prati, L. Catal. Lett. 2009, 133, 334.
18. Pawar, B.; Padalkar, V.; Phatangare, K.; Nirmalkar, S.;
Chaskar, A. Catal. Sci. Technol. 2011, 1, 1641.
19. Minkler, S. R. K.; Lipshutz, B. H.; Krause, N. Angew. Chem.
2011, 123, 7966.
20. Nishikata, T.; Lipshutz, B. H. Chem. Commun. 2009, 6472.
21. Saha, R.; Ghosh, A.; Saha, B. J. Coord. Chem. 2011, 64, 3729.
22. Lepiller, C. Direct Formic Acid Oxidation for Liquid-fed PEM Fuel Cells; Fuel cell Newsletter; Pragma Industries:
February 2012.
23. Kordesch, K. V.; Simader, G. R. Chem. Rev. 1995, 95, 191.
24. Yadav, M.; Singh, A. K.; Tsumori, N.; Xu, Q. J. Mater.
Chem. 2012, 22, 19146.
25. Boddien, A.; Mellmann, D.; Gärtner, F.; Jackstell, R.; Junge, H.; Dyson, P. J.; Laurenczy, G.; Ludwig, R.; Beller, M.
Science 2011, 333, 1733.
26. Fukuzumi, S.; Kobayashi, T.; Suenobu, T. J. Am. Chem.
Soc. 2010, 132, 1496.
27. Loges, B.; Boddien, A.; Junge, H.; Beller, M. Angew. Chem.
Int. Ed. 2008, 47, 3962.
28. Fellay, C.; Dyson, P. J.; Laurenczy, G. Angew. Chem. Int.
Ed. 2008, 47, 3966.
29. Samjeske, G.; Miki, A.; Ye, S.; Osawa, M. J. Phys. Chem.
B, 2006, 110, 16559.
30. Alexandris, P.; Hatton, T. A. Colloid Surf. 1995, 96, 1.
31. Islam, M.; Saha, B.; Das, A. K. J. Mol. Catal A: Chem.
2005, 236, 260.
32. Bayen, R.; Islam, M.; Saha, B.; Das, A. K. Carbohydr. Res.
2005, 340, 2163.
33. Islam, M.; Saha, B.; Das, A. K. Int. J. Chem. Kinet. 2006, 38, 531.
34. Mandal, J.; Chowdhury, K. M.; Paul, K.; Saha, B. J. Coord.
Chem. 2010, 63, 99.
35. Islam, M.; Das, A. K. Carbohydr. Res. 2008, 343, 2308.
36. Islam, M.; Das, A. K. Prog. React. Kinet. Mech. 2008, 33, 219.
37. Das, A. K. Inorg. React. Mech. 1999, 1, 161.
38. Das, A. K.; Das, M. J. Chem. Soc. Dalton Trans. 1994, 589.
39. Meenakshisundaram, S. P.; Gopalakrishnan, M.; Nagara- jan, S.; Sarathi, N. Catal. Commun. 2007, 8, 713.
40. Figgis, B. N. Introduction to Ligand Fields; Wiley East- ern Limited. New Delhi, India, 1966; p 222.
41. Khan, Z.; Ud-Din, K. Transition Met. Chem. 2002, 27, 832.
42. Islam, M.; Saha, B.; Das, A. K. J. Mol. Catal. A: Chem.
2007, 266, 21.
43. Jorgensen, C. K. Absorption Spectra and Chemical Bond- ing in Complexes; Pergamon Press Ltd: Oxford, London, 1964; p 290.
44. Saha, B.; Das, M.; Mohanty, R. K.; Das, A. K. J. Chin.
Chem. Soc. 2004, 51, 399.
45. Taboada, P.; Attwood, D.; Ruso, J. M.; Garcia, M.; Sarmiento, F.; Mosquera, V. J. Colloid Interface Sci. 1999, 220, 288.
46. Akhtar, F.; Hoque, M. A. J. Bangladesh Chem. Soc. 2006, 19, 88.
47. Myers, D. Surfaces, Interfaces and Colloids: Principles and Applications; VCH Publishers: New York, 1946.
48. Bhattacharya, S.; Kumar, V. P. Langmuir 2005, 21, 71.
49. Zakharova, L.; Valeeva, F.; Zakharov, A.; Ibragimova, A.;
Kudryavtseva, L.; Harlampipidi, H. J. Colloid Interf. Sci.
2003, 263, 597.
50. Svensson, R.; Pamedytyte, V.; Juodiatyte, J.; Makuska, R.; Morgenstern, R. Toxicology 200, 168, 251.
51. Mandal, J.; Chowdhury, K. M.; Paul, K. K.; Saha, B. Open Catal. J. 2008, 1, 1.
52. Khan, Z.; Raju, S. M.; Ud-Din, K. Transition Met. Chem.
2003, 28, 881.
53. Meenakshisundaram, S.; Markkandan, R. Transition Met.
Chem. 2004, 29, 308.
54. Basu, A.; Saha, R.; Mandal, J.; Ghosh, S. K.; Saha, B. J.
Biomed. Sci. Eng. 2010, 3, 735.
![Figure 1. Absorption spectrum of reaction mixture (after com- com-pletion of reaction): [formic acid] T = 75×10 −4 mol dm −3 , [Cr(VI)] T = 5×10 −4 mol dm −3 , [H 2 SO 4 ] T = 0.5 mol dm −3 , Temp = 30 o C (i) Unpromoted: (The spectrum of the chromic](https://thumb-ap.123doks.com/thumbv2/123dokinfo/5298919.154258/2.892.464.805.778.1010/figure-absorption-spectrum-reaction-mixture-reaction-unpromoted-spectrum.webp)
![Figure 3. Absorption spectrum of reaction mixture with and without promoter (in absence of substrate): (i) [Cr(VI)] T = 5×10 −4 mol dm −3 , [H 2 SO 4 ] T = 0.5 mol dm −3](https://thumb-ap.123doks.com/thumbv2/123dokinfo/5298919.154258/4.892.464.808.149.387/figure-absorption-spectrum-reaction-mixture-promoter-absence-substrate.webp)
![Figure 4-2. Schematic representation of partitioning of substrate and active oxidant [AO + = Cr(VI)-promoter complex] in (a) Cationic surfactant, (b) Anionic surfactant and (c) Neutral surfactant.](https://thumb-ap.123doks.com/thumbv2/123dokinfo/5298919.154258/6.892.84.809.169.321/schematic-representation-partitioning-substrate-cationic-surfactant-surfactant-surfactant.webp)
