The domestic strain of cauliflower mushroom, Sparassis latifolia JF02-06 (KACC 54766) was harvested from the larch tree of a local mountain in Jeonnam. The draft genome sequence of JF02-06 was generated by combining Oxford nanopore sequencing and Illumina MiSeq platform. Genome assembly resulted in 103 scaffolds, with a total size of 39.33 megabases (Mb) and G + C content of 45.74%. The annotated genomic sequence contains 13,038 gene models with an average gene length of 1,693 base pairs (bp) and average exon number of 5.6.
Genome analysis of JF02-06 did not show much difference with S. crispa, suggesting the two strains were very close with each other. The elucidation of S. latifolia genome will provide important data for evolutionary analysis of Sparassis species.
Keywords: Sparassis latifolia, cauliflower mushroom, genome sequence, Illumina MiSeq
Cauliflower mushroom (Sparassis latifolia, formerly known as S. crispa) is an edible and medicinal mushroom with significant role as various sources of health supplements, pharmaceutics,
cosmetics, and other biological substances (Chandrasekaran et al., 2011; Kimura, 2013). Recent morphological characteristics and phylogenetic study have refined this mushroom as S.
latifolia for Eastern Asian strains, rather than S. crispa for European and North American strains (Ryoo et al., 2013).
Despite its potential value as nutraceuticals and pharmaceuticals, there are scarce studies of S. latifolia characterized at the genome level. In this study, we report a draft genome sequence of Korean strain JF02-06 of S. latifolia.
S. latifolia JF02-06 (KACC 54766) was harvested from on the stump of dead larch tree (Larix kaempferi) of a local mountain in Jeonnam area (32.25°N, 127.54°E) in October, 2009. The mycelia were grown in the laboratory as described previously by Lee et al. (2015). Genomic DNA isolation from the mycelia was performed using the modified cetyl trimethyl- ammonium bromide (CTAB) method (Lee and Taylor, 1990).
NGS analysis of samples was performed by the Theragen Bio.
The presented genome sequence of S. latifolia JF02-06 was performed using a combination of Oxford nanopore sequencing platform GridION and Illumina MiSeq platform with a 350-bp paired-end library (Caporaso et al., 2012; Eisenstein, 2012).
Korean Journal of Microbiology (2020) Vol. 56, No. 3, pp. 315-317 pISSN 0440-2413
DOI https://doi.org/10.7845/kjm.2020.0057 eISSN 2383-9902
Copyright ⓒ 2020, The Microbiological Society of Korea
Draft genome sequence of cauliflower mushroom,
Sparassis latifolia JF02-06, isolated from Korean larch tree
Yuanzheng Wu
1,2, Hyun-Seok Kim
3, Deuk-Sil Oh
3, and Hyun-Jae Shin
1*
1
Department of Biochemical and Polymer Engineering, Chosun University, Gwangju 61452, Republic of Korea
2
Ecology Institute, Qilu University of Technology (Shandong Academy of Sciences), Shandong Provincial Key Laboratory of Applied Microbiology, Jinan 250103, P. R. China
3
Jeollanamdo Forest Resources Research Insitute, Naju 58213, Republic of Korea
대한민국 낙엽송에서 분리된 버섯균 Sparassis latifolia JF02-06의 게놈 서열 초안
오원정
1,2・ 김현석
3・ 오득실
3・ 신현재
1*
1
조선대학교 공과대학 생명화학고분자공학과,
2산동성과학원 생태연구소,
3전라남도 산림자원연구소
(Received June 22, 2020; Revised July 7, 2020; Accepted July 7, 2020)
*For correspondence. E-mail: [email protected];
Tel.: +82-62-230-7518; Fax: +82-62-230-7226
316
∙ Wu et al.미생물학회지 제56권 제3호
Long-read assembly was obtained from Oxford nanopore sequencing results using CANU v1.8, an OLC (Overlap Layout Consensus) assembly method (Koren et al., 2017). Short-read assembly was obtained from Illumina sequencing results using IDBA-UD, a DBG (de bruijn graph) algorithm, in order to decode genome that cannot be covered in error correction and Oxford nanopore assembly results (Peng et al., 2012). The two assemblies were merged with HaploMerger2 program, after which alignment of the Illumina sequencing results to the assembly results and error correction were performed using the pilon tool (Huang et al., 2012). Then ultra highly conserved genes were identified using CEGMA and universal single orthologous genes were identified using BUSCO software to evaluate the assembly results (Parra et al., 2007; Simão et al., 2015).
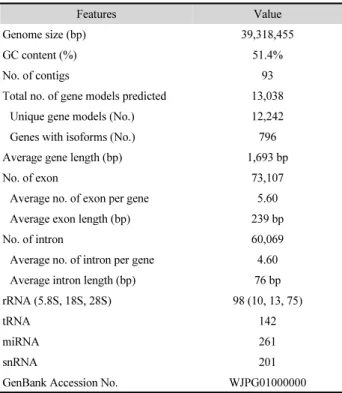
As shown in Table 1, a total of 39.3 megabases (Mb) clean genome-sequencing data was obtained by assembling appro- ximately 13.51 and 10.24-Gbp reads, respectively (> 500 × coverage). The draft genome consisted of 93 contigs with N50 length of 1.596 Mb and has 51.4% G + C content. The S.
latifolia JF02-06 genome in this study is of a similar size with Japanese strain S. crispa Scrmy26 (39.02 Mb), but quite different with previously reported Chinese strain S. latifolia Minxiu NO.1 (48.13 Mb) (Kiyama et al., 2018; Xiao et al., 2018). This
is probably due to the depth of sequencing, as revealed in the scaffold/contig numbers and N50 values employed by different sequencing methods.
Genome annotation was performed using BRAKER2 pipeline with AUGUSTUS tool for predicting protein coding genes (Hoff et al., 2016). The assembled genomic sequence of JF02-06 was annotated and 13,038 gene models including the coordinates of each intron and exon, as well as the beginning and end of the transcript were obtained, characterized by an average gene length of 1,693 bp and average exon number of 5.6 (Table 1).
The genes predicted formed transcripts with an average length of 1.45 kb and proteins with an average length of 484.67 amino acids. The genome also contained coding sequences of 98 rRNAs, 142 tRNAs, 261 miRNAs, and 201 snRNAs. The number of genes in the genome of JF02-06 was comparable with that in genomes of Scrmy26 and Minxiu NO.1 (13,157 and 12,471 genes, respectively).
The genomic data of S. latifolia JF02-06 will provide helpful information to expand academic diversity of mycological research.
Nucleotide sequence accession number
The draft genome sequence of Sparassis latifolia JF02-06 (KACC 54766) has been deposited in GenBank under the accession number WJPG01000000 (https://www.ncbi.nlm.nih.
gov/nuccore/WJPG00000000).
적 요
담자균의 일종으로 꽃송이버섯으로 알려진 Sparassis latifolia JF02-06 (KACC 54766)이 전라남도 구례군의 산림지역 낙엽 송에서 채집, 분리되었다. JF02-06의 게놈 서열은 Oxford nano- pore sequencing과 Illumina MiSeq platform 분석을 통해 작성 되었으며, 게놈 조립으로 총 크기는 39.33 Mb이고 G + C 함량 은 45.74%인 103개의 스캐폴드가 생성되었다. 주석이 달린 게놈 서열은 평균 유전자 길이가 1,693 bp이고 평균 엑손수가 5.6인 13,038개의 단백질 코딩 유전자를 포함하고 있다. 본 S.
latifolia 게놈 서열 초안은 식의약용 버섯인 Sparassis 종의 진 화 분석에 중요한 데이터를 제공할 것이다.
Table 1. Genome features of Sparassis latifolia JF02-06
Features Value
Genome size (bp) 39,318,455
GC content (%) 51.4%
No. of contigs 93
Total no. of gene models predicted 13,038
Unique gene models (No.) 12,242
Genes with isoforms (No.) 796
Average gene length (bp) 1,693 bp
No. of exon 73,107
Average no. of exon per gene 5.60
Average exon length (bp) 239 bp
No. of intron 60,069
Average no. of intron per gene 4.60
Average intron length (bp) 76 bp
rRNA (5.8S, 18S, 28S) 98 (10, 13, 75)
tRNA 142
miRNA 261
snRNA 201
GenBank Accession No. WJPG01000000
Genome sequence of Sparassis latifolia JF02-06∙
317
Korean Journal of Microbiology, Vol. 56, No. 3
Acknowledgments
This research was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) [No. NRF-2017R1A2B4006204]. Dr.
Yuanzheng Wu thanks Shandong Provincial Key Research and Development Program (International Science and Technology Cooperation) [No. 2019GHZ033] and International Science and Technology Cooperation Program of Shandong Academy of Sciences [No. 2019GHPY05] for financial support.
References
Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, Owens SM, Betley J, Fraser L, Bauer M, et al. 2012.
Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 6, 1621–1624.
Chandrasekaran G, Oh DS, and Shin HJ. 2011. Properties and potential applications of the culinary-medicinal cauliflower mushroom, Sparassis crispa Wulf.:Fr. (Aphyllophoromycetideae): a review.
Int. J. Med. Mushrooms 13, 177–183.
Eisenstein M. 2012. Oxford nanopore announcement sets sequencing sector abuzz. Nat. Biotechnol. 30, 295–296.
Hoff KJ, Lange S, Lomsadze A, Borodovsky M, and Stanke M. 2016.
BRAKER1: unsupervised RNA-Seq-based genome annotation with GeneMark-ET and AUGUSTUS. Bioinformatics 32, 767–
769.
Huang S, Chen Z, Huang G, Yu T, Yang P, Li J, Fu Y, Yuan S, Chen S, and Xu A. 2012. HaploMerger: reconstructing allelic relationships for polymorphic diploid genome assemblies. Genome Res. 22, 1581–1588.
Kimura T. 2013. Natural products and biological activity of the pharmacologically active cauliflower mushroom Sparassis crispa.
BioMed Res. Int. 2013, 982317.
Kiyama R, Furutani Y, Kawaguchi K, and Nakanishi T. 2018.
Genome sequence of the cauliflower mushroom Sparassis crispa (Hanabiratake) and its association with beneficial usage.
Sci. Rep. 8, 1–11.
Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, and Phillippy AM. 2017. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res.
27, 722–736.
Lee DJ, Jang MC, Jo AR, Choi HJ, Kim KS, and Chi YT. 2015. Noble strain of Sparassis latifolia produces high content of β-glucan.
Asian Pac. J. Trop. Biomed. 5, 629–635.
Lee SB and Taylor JW. 1990. Isolation of DNA from fungal mycelia and single spores. In Innis MA, Gelfand DH, Sninsky J, and White TJ. (eds.). PCR protocols. pp. 282–287. Academic Press.
San Diego, California, USA.
Parra G, Bradnam K, and Korf I. 2007. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23, 1061–1067.
Peng Y, Leung HC, Yiu SM, and Chin FY. 2012. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28, 1420–1428.
Ryoo R, Sou HD, Ka KH, and Park H. 2013. Phylogenetic relationships of Korean Sparassis latifolia based on morphological and its rDNA characteristics. J. Microbiol. 51, 43–48.
S imão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, and Zdobnov EM. 2015. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212.
Xiao D, Ma L, Yang C, Ying Z, Jiang X, and Lin YQ. 2018. De novo sequencing of a Sparassis latifolia genome and its associated comparative analyses. Can. J. Infect. Dis. Med. Microbiol. 2018, 1857170.