기획특집 바이오 인포메틱스-
비교 모델링 방법을 이용한 단백질 구조 예측
유 아 림․이 원 태*․양 대 륙† 고려대학교 화공생명공학과, *연세대학교 생화학과
Protein Structure Prediction Using Comparative Modeling Method
Ahrim Yoo, Weontae Lee*, and Dae Ryook Yang† Department of Chemical and Biological Engineering, Korea University
*Department of Biochemistry, Yonsei University
Abstract: 생물 정보학은 생명 공학과 정보 기술을 활용하여 생명체가 지닌 막대한 양의 정보를 수집하고 이를 분석하 여 유용한 정보를 얻어내고자 하는 연구 분야이다. 많은 실험 연구자들에 의하여 단백질을 구성하는 아미노산 서열 정 보와 구조 정보들이 데이터베이스에 저장된다. 핵자기공명법(NMR)과 X선결정결정법 등의 실험에 의한 단백질 3차원 구조 결정 방법은 결과의 정확성에 비하여 많은 시간과 비용이 요구된다. 그러므로 단백질 1차 구조 정보(아미노산 서 열)만을 이용하여 단백질 3차원 구조를 예측하는 연구가 활발하게 진행 중이다. 구조 예측 방법들은 단백질을 구성하는 아미노산 서열이 이미 구조가 실험으로 결정된 단백질들의 아미노산 서열과의 유사성 정도에 따라 비교 모델링 방법 (comparative modeling), 구조 인식 방법(fold recognition), 새 구조 방법(new fold) 세 가지로 분류된다. 이 글을 통하 여 단백질 구조 예측 방법들과 모델링된 단백질 3차원 구조를 이용하여 그 기능을 분석하는 방법에 대하여 소개하고자 한다.
Keywords: protein structure prediction, comparative modeling method, fold recognition method, new fold method, CASP, ACC oxidase, docking
1. 서 론1)
생물 정보학은 생명 공학과 정보기술을 활 용하여 생명체가 지닌 막대한 양의 정보를 수 집하고 이를 분석하여 유용한 정보를 얻어내 고자 하는 연구 분야이다. 많은 연구자들에 의 하여 유전자 서열, 단백질 서열, 단백질 구조, 생화학 반응의 물질 경로와 상호 작용 등에 관련된 다양한 데이터베이스들이 구축되고 있 다[1]. 이중 PDB (Protein Data Bank)는 실 험과 모델링 방법을 이용하여 결정된 단백질 의 3차원 구조 정보가 저장되어 있다(http://
www.rcsb.org). 20개의 아미노산(amino acid)
† 주저자 (E-mail: [email protected])
의 조합으로 이루어진 단백질은 생명 활동의 주요 매개체이다. 단백질은 생물체 내에서 몸 의 구성 성분, 세포내 생화학 반응의 촉매, 각종 신호를 전달하는 센서 등의 역할을 하고 있다.
그러므로 생명현상을 이해하기 위해서는 단백질 의 기능을 알아내는 것이 꼭 필요하다[2].
단백질은 생물학적 기능을 수행하기 위해 접힘(folding)을 통하여 고유한 3차원 구조를 가져야 한다. 단백질의 3차원 구조가 결정이 되 면 이를 이용하여 대상 단백질의 활성 사이트 나 다른 단백질 또는 분자들과의 상호작용에 대한 예측이 가능해진다. 그러므로 대상 단백 질의 3차원 구조를 규명하는 것은 그 기능을 밝히기 위한 연구의 출발점이라고 할 수 있다.
구조를 결정하기 위한 실험 과정은 대상 단백
질을 순수하게 분리 정제하여 시료를 준비한 후, 핵자기공명법(Nuclear Magnetic Resonance:
NMR)이나 X선결정법(X-ray crystallography) 을 이용하여 데이터를 얻어 이를 분석하는 것 이다. 위 두 방법의 차이는 시료의 상태가 핵 자기공명법의 경우 수용액 상태인 반면 X선 결정법의 경우 단결정 상태라는 것이다. 핵자 기공명법은 수용액 상태의 시료를 이용하기 때문에 실제 생체 내에서 단백질이 가지고 있 는 유사한 구조를 얻을 수 있으며 단백질의 backbone 및 side-chain의 동역학적 특징을 관측할 수 있다는 장점이 있다. 하지만 측정 가능한 분자량에 제한(100 kDa 이하)이 있으 며 결과 분석에 시간이 오래 걸린다. X선결정 법은 단결정상태로 실험을 하기 때문에 대상 단백질에 따라 결정화의 어려움이 있는 경우 도 있으며 동역학적 특징은 관찰할 수 없으나, 단백질 크기에 제한이 없으며 일단 단결정 시 료가 준비되면 결과 분석 속도가 상대적으로 빠른 장점을 가지고 있다. 위 두 방법은 단백 질 구조 결정 분야의 독보적인 위치를 차지하 고 있다[3,4].
하지만 실험을 통한 방법에는 많은 시간과 비용이 소모되며, 단백질 특성에 따라 분리 정 제의 어려움 때문에 대상 선택에 제약이 있다.
또한 2006년 현재 결정된 단백질 3차원 구조 의 개수는 38,479개임에(http://www.ncbi. nlm.
nih.gov/Genbank/genbankstats.html) 비하여 단 백질의 1차 구조(아미노산 서열)가 실험으로 결정된 개수 52,016,762개이다(http://www.rcsb.
org). 즉 DNA sequencing에 의해 아미노산 서열을 결정하는 실험의 속도가 구조 결정 실 험보다 훨씬 더 빠르다. 그러므로 단백질을 구 성하는 아미노산 서열의 정보를 이용하여 컴 퓨터 계산으로 3차원 구조를 결정하는 구조 예측 방법을 개발하는 연구가 활발하게 진행 중이다. 기존에 개발된 방법들은 비교 모델링 방법(comparative 또는 homology modeling), 구조 인식 방법(fold recognition), 새 구조 (new fold 또는 ab initio) 방법 세 가지로 분류 될
수 있다[5,6]. 이 글을 통하여 먼저 세 가지 단백질 구조 예측 방법과 최근 동향을 정리하 고, 비교 모델링 방법에 대한 예제로 식물 성 장 호르몬인 에틸렌 합성에 효소로 작용하는 1-aminocyclopropane-1-carboxylate oxidase (ACC oxidase)의 구조를 예측하는 과정을 소개하고 자 한다.
2. 단백질 구조 예측 방법
단백질 구조 예측이란 이론식을 이용하여 단백질의 1차 구조(아미노산의 서열)로부터 단백질의 3차 구조를 결정하는 것을 뜻한다 [5]. 3차 구조란 단백질이 생물학적 기능을 할 수 있게 되는 고유 구조이며, 이 구조는 대상 단백질의 기능 규명, 신소재 및 의약품 설계 등과 같은 연구에 이용될 수 있다. 기존에 개 발된 방법은 구조 데이터베이스(PDB) 내의 단백질들과 대상 단백질의 아미노산 서열간의 유사도(similarity)에 따라 다음 세 가지로 분 류된다[6,7].
첫 번째 방법은 비교 모델링 방법(comparative or homology modeling)이다. 비교 모델링 방 법은 “유사한 아미노산 서열을 가진 단백질들 은 서로 유사한 3차원 구조를 가질 것이다”라 는 가정에서 출발한다. 대상 단백질(target)의 아미노산 서열과 유사한 서열을 가지는 단백 질(template: 주형)이 PDB 내에 존재할 경우 (즉, 3차원 구조가 알려졌을 때), 주형 단백질 의 구조를 이용하여 대상 단백질의 구조를 계 산 하는 방법이다. 이 방법은 두 단백질 사이 의 아미노산 서열의 유사도가 30% 정도 이상 의 경우 적용이 가능하고 유사도가 높을수록 결과가 정확해진다. 대상 단백질의 구조를 결 정하는 단계는 먼저 구조 데이터베이스(PDB) 를 이용하여 주형 단백질을 검색한 후, 주형 단백질의 구조 정보(원자들 사이의 거리, 각도 등)를 측정하여, 이 정보를 구속 조건(restraint) 으로 사용하여 대상 단백질의 삼차 구조를 만
들어내는 것이다. 이 방법을 restraint-based method라고 하며, 현재 가장 많이 이용되고 있는 프로그램인 MODELLER (http://www.
salilab.org/modeller/)에 적용되어 있다[8]. 다른 방법으로는 fragment-based method라는 것이 있는데, 이는 대상 단백질의 아미노산 서열을 주형과 서열이 잘 일치하는 conserved region 과 잘 일치하지 않는 variable region으로 구분 한 후, 먼저 conserved region의 구조를 만든 다. 주로 conserved region에 해당하는 것들은 단백질의 2차 구조(α-helix, β-strand)들이 된 다. 주형 단백질의 구조를 이용하여 α-helix와 β-strand들의 공간상 배향을 결정하고 이들 사 이를 연결하는 loop 구조를 만들어 낸다. Loop 들은 주로 variable region에 해당된다. 이 방 법이 적용된 프로그램으로는 COMPOSER와 웹서버 형태로 제공되는 SWISS-MODEL (http://swissmodel.expasy.org/)이 있다[5,6].
두 번째 방법은 구조 인식 방법(fold recog- nition)이다. PDB 내의 단백질들을 구조 유형 별로 분류한 데이터베이스인 SCOP (Structural Classification of Proteins: http://scop.berkeley.
edu)과 CATH (Class, Architecture, Topology, Homologuous superfamily: http://cathwww.
biochem.ucl.ac.uk/latest/index.html)를 보면 아 미노산 서열간의 유사성은 보이지 않지만 구 조적으로 유사한 단백질들이 관찰된다. 이러한 단백질은 remote homology 또는 distance ho- mology 관계에 있다고 한다. 구조 인식 방법 의 기본 가정은 “새롭게 발견된 단백질은 구 조 데이터베이스 안에 있는 단백질들과 서열 이 유사하지 않더라도 구조적으로 비슷할 가 능성이 있다”는 것이다[6]. 구조 인식 방법의 주된 과제는 remote homology를 가지는 주형 을 어떻게 찾아내느냐이다. 이 방법들 중 하나 가 threading 방법이다. Threading 방법을 적 용하기 위해서는 구조가 알려진 단백질들 중 대표 구조들만으로 구성된 구조 라이브러리 (fold library)와 점수 함수(score function)가 필요하다. 아미노산 서열이 주어져있는 대상
단백질의 구조를 구조 라이브러리에 있는 모 든 형태로 만들어 본 후, 점수 함수를 이용하 여 총 점수를 계산한다. 그 중에서 가장 좋은 점수를 가지는 구조를 주는 라이브러리 구조 를 주형 단백질로 선택한다. 주형 단백질이 선 택되면 MODELLER 등을 이용하여 대상 단 백질의 구조를 만들 수 있다. 이 방법이 적용된 대표적인 프로그램은 Jones그룹(UCL, London) 에서 개발한 PSIPRED (http://bioinf.cs.ucl.ac.uk/
psipred/)이며 웹에서 서비스된다. 주형 단백 질을 결정하기 위하여 threading 방법은 3차원 구조 라이브러리를 이용하는 반면, 프로파일 (profile)이란 아미노산 서열만을 가지고 계산 한 점수 행렬을 이용하는 방법도 있다. 이 방 법을 프로파일-프로파일 정렬(profile-profile align- ment)이라고 하며, 3차원 구조 정보를 이용하 지 않기 때문에 구조 라이브러리를 검색하는 데 소비되는 시간을 절약할 수 있다. 프로파일 이란 PSI-BLAST라는 프로그램에 의해 계산 된 위치에 의존하는 PSSM (Position-Specific Scoring Matrix)이라는 점수행렬이며, 이 PSSM 을 이용하여 대상 단백질과 3차원 구조를 알 고 있는 단백질의 PSSM을 정렬하여 주형 단 백질을 찾아내는 것이 프로파일-프로파일 정 렬 방법이다. FFAS03 (Fold and Function Assignment System: http://ffas.ljcrf.edu/
ffas-cgi/cgi/ffas.pl)과 SAM-T02 (HMM-based Protein Structure Prediction: http://www.cse.
ucsc.edu/compbio/HMM-apps)이란 프로그램 이 이 방법이 적용된 것이다. PSSM은 PSI- BLAST (http://www.ncbi.nlm.nih.gov/blast/) 를 이용하여 직접 계산 할 수도 있으며, 프로 파일 데이터베이스인 PSPDB (Protein Sequence Profile Data Bank: http://www.cheric.org/
research/pspdb)에 이미 계산된 값을 이용할 수도 있다[6].
세 번째 방법은 새 구조 방법(New Fold method)이다. 위의 두 방법은 구조 데이터베 이스에 존재하는 주형 단백질의 구조를 이용 하여 대상 단백질의 구조를 만들어내는 방법
이었다. 하지만 remote homology 관계있는 단 백질조차도 검색되지 않는 새로운 구조(New fold)의 경우, 초기에는 주로 이론식에 근거한 방법(ab initio)을 이용하여 구조를 예측하였다.
ab initio 방법이란 퍼텐셜 에너지(Potential Energy) 함수를 이용하여 단백질 구조의 에 너지 함수를 표현하고 분자 동역학(Molecular Dynamics), 몬테칼로 방법(Monte Carlo method), 광역 최적화(Global optimization)의 계산을 통 하여 3차 구조를 결정하는 방법이다. 하지만 퍼텐셜 에너지가 최소가 되는 구조를 결정하 기 위해서는 많은 시간과 엄청난 계산 능력이 요구되어지기 때문에 적용 가능한 단백질의 크기가 한정적이며, 퍼텐셜 에너지 함수 자체 의 부정확성 때문에 결과의 정확성이 보장되 지 않는다. 이 방법은 흥미로운 연구 소재를 많이 가지고 있기 때문에 최적화 계산 방법이 나 퍼텐셜 함수 개선에 대한 활발한 연구가 진행 중이다[6]. 최근 조각 맞추기 방법 (Fragment Assembly)이라는 실용적인 방법 이 제안되었다. 조각 맞추기 방법의 기본 가정 은 “새로운 구조를 가진 단백질이더라도 이를 구성하는 부분의 모양은 이미 구조가 알려진 단백질들의 한부분과 유사하거나 동일할 것이 다”라는 것이다. ab initio 방법과는 다르게 퍼 텐셜 함수에만 의존하는 것이 아니라, 조각 라 이브러리(Fragment Library)라는 데이터베이 스가 이용된다. 먼저 PDB에 있는 모든 단백 질들의 구조들의 PSSM을 계산한 후, 이 점수 행렬을 일정한 조각(예를 들면 아미노산 3개 해당하는 길이)을 나누어 조각 라이브러리를 구성한다. 대상 단백질이 주어지면 라이브러리 구성 할 때와 동일하게 PSSM을 계산하여 조 각 라이브러리와 같은 크기로 나눈 다음 조각 라이브러리에 있는 데이터와 비교하여 유사한 PSSM을 가진 조각들을 추려낸다. 이렇게 추 려진 조각들이 대상 단백질을 구성하는 각 조 각들의 후보가 된다. 이때 퍼텐셜 에너지 함수 를 이용하여 각 후보들 중에서 최소의 퍼텐셜 에너지를 가지는 조각들의 조합을 찾아내어
최종 구조를 결정한다. 그러므로 이 방법은 조 각 라이브러리를 이용하는 생물정보학 요소와 퍼텐션 에너지 함수를 이용하는 ab intio 요소 를 동시에 가지고 있기 때문에 ab intio 방법 에 비하여 효율적이며 예측 결과의 정확도도 높 은 편이다. 또한 구조 인식 방법에도 적용이 가 능하다. Baker 그룹(University of Washington, Seattle)에서 개발한 Robetta (http://robetta.
bakerlab.org/)라는 구조 예측 서버에 이 방법 이 적용되었다[6].
3. CASP (community-wide experiment on the Critical Assesment of tech- niques for protein Structure Pre- diction)
CASP은 기존에 개발된 구조 예측 방법들의 성능을 평가하기 위한 대회이다. 1994년 미국 생 명고등연구센터의 J. Moult에 의해 시작되었다.
이후 2년마다 주최 측(http://predictioncenter.
org/)이 전 세계의 실험 그룹들로부터 곧 3차 구조가 결정될 단백질들의 서열을 제공받아 문제를 제출하고, 참가 그룹들은 대회 기간 동 안 각 그룹들이 개발한 방법을 이용하여 대상 단백질들의 구조를 예측하여 정해진 시간 내 에 주최 측에 결과를 제출한다. 구조 예측에 주어진 문제들은 비교 모델링(CM), 구조 인 식(FR), 새 구조 방법(NF) 3가지 분야로 구 분된다. 이 대회를 통하여 구조 예측 분야의 다양한 접근법과 방법들의 장단점이 알려졌다.
2004년에 개최되었던 CASP6는 모두 25개국 에서 200여 그룹이 참여하였다. 잘 알려진 바 와 같이 CM 분야의 성능은 대상 단백질과 주 형 단백질 사이의 아미노산 서열 일치도 정도 에 크게 의존한다. 서열 정렬시 메타 프로파일 정렬 방법을 이용한 Ginalski 그룹(University of Texas, Dallas)의 Meta-Basic 방법이 가장 좋은 성과를 보였다[9]. FR분야는 데이터베이
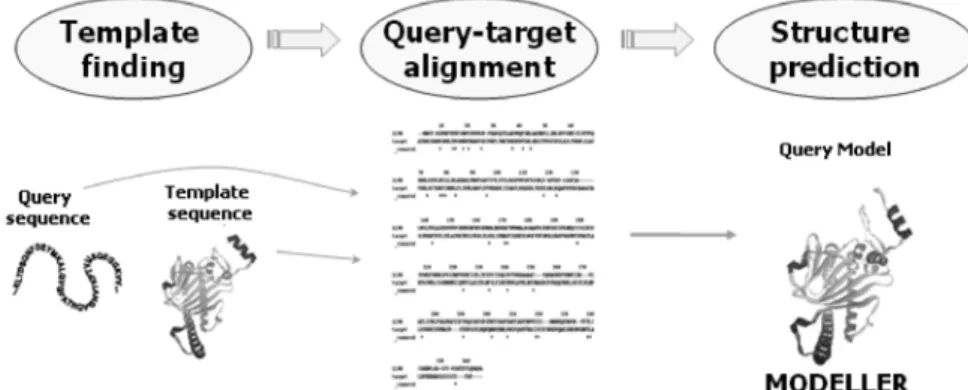
Figure 1. 비교 모델링 방법을 이용한 단백질 구조 예측.
스에서 주형 단백질의 검색의 가능성 여부에 따라, FR/H (Fold Recognition/Homology)와 FR/A (Fold Recognition/Analogous) 분야로 나뉘어 평가되었다. FR/H 분야에서는 CM에 서와 마찬가지로 대상 단백질과 주형 단백질 사이의 아미노산 서열 정렬에서 좋은 성능을 보이는 방법을 개발한 Ginalski 그룹이 제일 좋은 성과를 보였다. FR/A과 NF 분야에서는 조각 맞추기 방법을 도입하여 매 대회마다 좋 은 결과를 기록하고 있는 Baker그룹(Univer- sity of Washington, Seattles)의 결과가 다른 그룹의 결과들에 비하여 월등히 좋았다. 또한 이 그룹이 개발한 구조 예측 서버인 Robetta 도 10권 안에 순위를 기록하여, 이들이 적용한 조각 맞추기 방법이 구조 예측에 효과적인 방법 임을 대회를 통하여 보여주었다[10,11]. CASP6 의 결과, 새 구조 방법의 연구에 적용된 조각 맞추기 방법이 remote homology를 검색하는 기 능 개선에 영향을 주어, 구조 인식 방법의 성능 을 월등하게 개선시켰다. 또한 CM과 FR/H 분 야의 문제들에 사용된 방법들과 FR/A과 NF 분야에 적용되는 방법들에서 각각 그 경계선 들이 모호해졌다[12]. 2006년 CASP7 대회가 시작되었으며, 9월 현재 참가 그룹들은 결과 제출을 끝내고 9월에 발표될 평가 결과를 기다 리고 있는 중이다. 앞에서 지적한대로 각 방법 들의 경계가 모호해졌기 때문에, 이번 대회부 터 문제들이 “template based modeling”과 “tem-
plate free modeling" 두 분야로만 구분되어 출 제되었다[13]. CASP 대회의 결과는 Proteins:
Structure, Function, and Bioinformatics라는 학술지의 특별호로 발표된다.
4. 비교 모델링 방법을 이용한 효소 구조 예측
식물 생장 호르몬인 에틸렌 생성에 관여하 는 ACC oxidase의 구조 예측하는 방법을 예 제로 하여 비교 모델링 방법과 모델 구조가 어떻게 이용되는지 소개하고자 한다. 대상 단 백질은 사과(Malus domestica Borkh)에서 에 틸렌을 생합성하는 최종 단계인 1-aminocyclo- propane-1-carboxylate의 산화반응에 관여하는 효소로 non-heme iron enzyme 패밀리에 속한 다. 본 연구 그룹에서는 이 효소의 구조를 비 교 모델링 방법을 이용하여 예측하였으며, 그 구조를 이용하여 효소의 활성화 사이트의 특 성을 분석하였다[14].
Figure 1에서 보여주듯이 비교 모델링 단계 는 다음과 같다.
1) 주형 단백질 검색
2) 주형 단백질과 대상 단백질의 아미노산 서열 정리
3) 대상 단백질의 구조 생성
첫 번째 단계로 주형 단백질을 검색하기 위
(A) (B)
Figure 2. 모델링에 이용된 주형 단백질(A)과 ACC oxidase의 구조 예측 결과(B).
하여 BLAST나 PSI-BLAST를 이용하여 아 미노산 서열 데이터베이스(PDB)를 검색한다.
이 검색 프로그램을 이용하기 위해서는 NCBI (National Center for Biotechnology Information:
http://www.ncbi.nlm.nih.gov/BLAST/)의 홈 페이지에서 다운을 받거나 온라인상에서 직접 사용이 가능하다. BLAST의 경우 유사한 서 열을 찾기 위해 BLOSUM62라는 점수 행렬을 사용하는 반면 PSI-BLAST (PSI는 Position- Specific Iterated를 뜻한다)는 프로파일이란 서열에 의존하는 점수행렬(PSSM)을 이용한 다. 이 PSSM을 이용할 경우, 앞에서 언급했 듯이 remote homology 관계의 주형 단백질 검색에 좋은 성능을 보인다[6]. BLAST 검색 결 과 페튜니아(Petunia hybrida)의 ACC oxidase (PDB ID: 1WA6)가 발견되었다. 이 주형 단 백질은 대상 단백질과 같은 기능을 하는 효소 일 뿐만이 아니라, 두 단백질간의 아미노산 서 열이 76.8% 정도 일치하였다. 비교 모델링 방 법의 경우, 서열간의 유사도가 높을수록 그 결 과의 신뢰도가 높아지기 때문에 이 주형 단백 질의 구조를 이용하여 대상 단백질의 구조를 쉽게 결정 할 수 있다.
두 번째 단계는 주형 단백질과 대상 단백질 사이의 아미노산 서열 정렬이다. ACC oxidase 의 경우 주형 단백질과 매우 높은 유사도를
가지고 있기 때문에 BLAST에서 제공하는 아 미노산 서열 정렬의 결과를 그대로 이용하였 다. 만약에 서열간의 유사도가 25~30% 정도 라면 서열 정렬의 문제가 쉽지 않게 된다. 이 경우 PSSM을 이용하여 서열을 정렬하는 것 이 더 좋은 결과를 얻을 수 있다.
세 번째 단계는 단백질 3차원 구조의 생성이다.
이 때 MODELLER가 이용된다. MODELLER는 Sali 그룹(University of California, San Fran- cisco)에서 개발한 대표적인 비교 모델링 프로 그램으로 주형 단백질의 원자들 사이의 거리, 각도 등 구조 정보로부터 얻은 공간적 구속 조건(spatial restraints)을 이용하여 퍼텐셜 함 수를 만들고 이를 만족하는 대상 단백질의 구 조를 모델링한다. 계산을 위한 입력 값은 주형 단백질의 구조와 서열 정렬 결과이며 결과 구 조는 PDB 형식으로 제공해준다. 이 프로그램 은 구조 예측 외에도 서열 정렬, 구조 정렬, 데이터베이스 검색 등 여러 가지 기능이 가능 하다. Figure 2는 모델링에 사용된 주형 단백 질(A)과 대상 단백질의 예측 결과 구조(B)를 보여준다.
이와 같은 방법으로 결정된 효소의 구조는 기능 해석 즉 활성화 자리의 특성 분석에 이 용이 될 수 있다. ACC oxidase가 에틸렌의 합 성을 하기 위해서는 반응물인 ACC, dioxygene, 조효소(cosubstrate)인 ASC (ASCorbate), 보 조인자(cofactor)인 철 이온 등과 결합해야 한 다. 비교 모델링 방법의 단점은 계산 결과가 주형 단백질의 구조에 전적으로 의존한다는 것 이다. 주형 단백질인 페튜니아의 ACC oxidase 의 구조는 철 이온만 결합되어 있는 형태로, 반 응물이 결합되지 않은 비활성화 상태의 구조 이기 때문에 활성화된 모델 구조를 얻기 위해 서는 구조의 개선이 필요하다. 개선을 위하여 데이터베이스에서 추가로 사용 가능한 주형 단백질을 검색하였다. 그 결과 같은 non-heme iron enzyme 패밀리에 속하는 IPNS (Isopenicilin N synthase)가 검색되었다. IPNS의 경우, 철 이 온(cofactor) 결합구조(PDB ID: 1IPS), diox-
(A) (B)
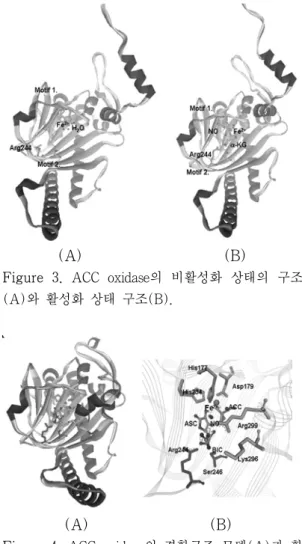
Figure 3. ACC oxidase의 비활성화 상태의 구조 (A)와 활성화 상태 구조(B).
(A) (B)
Figure 4. ACC oxidase의 결합구조 모델(A)과 활 성화 사이트(B).
ygene의 유사물인 NO와의 결합구조(PDB ID:
1BLZ), 반응물 결합 구조(PDB ID: 1BKO) 등 모두 세 가지 형태의 실험 구조가 발표되 었다. 이 구조들을 비교해본 결과, 반응물의 결합 유무에 따라 활성화 자리에 있는 side- chain의 방향이 달라지는 것이 관찰되었다. 그 러므로 반응물이 결합된 IPNS의 활성화 사이 트 구조를 주형으로 추가하여 모델을 개선 할 수 있었다. MODELLER의 장점은 다른 화합 물이 결합된 형태의 주형 구조를 이용한다면 화합물과 결합된 형태의 단백질 구조도 모델 링 할 수 있는 것이다. 그러므로 IPNS/철이온
/NO의 결합구조(PDB ID:1BLZ)의 활성화 사 이트 부분구조와 같은 패밀리 효소들이 반응 시 공통적으로 요구되어지는 NO와 철 이온을 주형으로 추가하여 ACC oxidase/철이온/NO 의 결합 구조를 Figure 3과 같이 모델링 하였 다. 철 이온과 결합하는 잔기들은 His177-X- Asp179-X(53)-His234들로 ACC oxidase와 주 형 단백질들이 속한 non-heme iron enzyme 패밀리에서 그 구조적 특성이 잘 보존되어 있 다. 모델링 결과 non-heme iron enzyme 패밀 리들의 공통 구조인 철 이온 결합 구조(Motif 1)가 잘 보존되고 있음을 확인하였다. Figure 3의 A와 B에서 보여 주듯이 Motif 2의 244번 잔기(Arg)가 ACC oxidase에 inhibitor인 α- KG가 존재할 경우, 방향이 활성화 사이트 쪽 으로 위치가 변동한 것을 확인 할 수 있었다.
Motif 2는 반응의 조효소인 ASC와 결합 할 것으로 예상되는 잔기들(Arg244-X-Ser246)을 의미한다.
하지만 대상 효소의 기능 해석에서 가장 중 요한 반응물(ACC)과 조효소(ASC)와의 결합 은 ACC oxidase의 고유한 특징이므로, 이 결 합 구조에 대한 주형 단백질을 찾을 수 없다.
그러므로 본 연구에서는 ACC와 ASC가 효소 에 결합된 구조를 모델링하기 위하여 docking 방법을 적용하였다. Docking이란 단백질과 작 은 분자(ligand)간의 결합 에너지를 계산하여, 단백질 내에서 작은 분자들이 어떤 위치에 있 을 때 결합 구조가 가장 안정화되는지를 찾는 방법이다. Olson 그룹에서 개발한 Autodock4.0 이란 프로그램(http://www.scripps.edu/mb/olson/
doc/autodock/)을 이용하여 다음 Figure 4와 같은 최종 구조를 예측하였다.
이러한 단계를 거쳐 모델링된 결합 구조를 검증 하는 방법 중 하나로 site-directed mutagenesis란 실험이 있다. 이 실험은 단백질을 구성하는 잔기 (residue)중 하나를 다른 종류의 아미노산으로 치환한 후, 효소의 활성에 영향이 있는가를 확 인하는 방법이다. 만약 치환 결과 효소의 활성 이 감소하면 그 잔기는 활성화에 중요한 역할
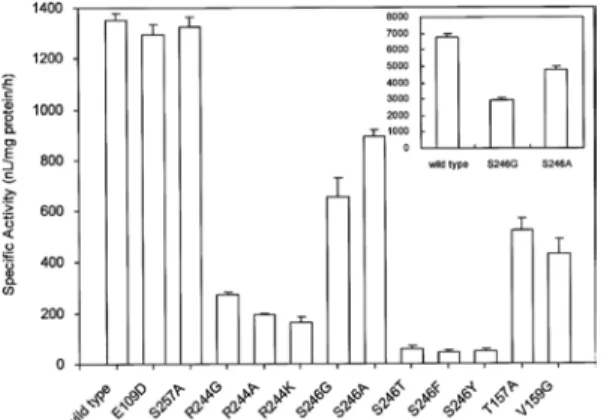
Figure 5. Wild type과 치환된 효소들의 specific activity 비교 실험 결과.
을 하는 것이며, 치환한 아미노산의 물리화학 적 특성에 따라 활성화 자리에서 어떤 기능을 하는지 분석도 가능하다. 결합 구조 모델링을 통하여 ASC는 ACC oxidase의 Arg244번과 Ser246번과 결합하고 있다는 것을 알 수 있었 다. 이를 확인하기 위하여 두 잔기들을 각각 Gly, Ala, Lys 등으로 치환 후, Figure 5와 같 은 활성의 변화 결과를 얻었다. 실험 결과 244 번 잔기의 경우 크기, 정전기적 상호작용 (electrostatic interaction) 등의 영향을 살펴본 결과, 모두 활성에 영향이 있었다. 하지만 246 번의 경우 수소결합이나 정전기적 상호작용에 대한 영향을 없었으나 크기가 큰 아미노산으 로 환한 경우 활성이 감소하는 것을 확인할 수 있었다. 그러므로 이 두 잔기들이 ASC와 효소의 결합에 관여하는 것을 구조 모델링과 site directed mutagensis 실험을 통하여 확인 할 수 있었다[14,15].
5. 결 론
이 글을 통하여, 단백질 구조 예측 방법과 예측된 3차원 구조를 이용하는 방법에 대하여 살펴보았다. ACC oxidase라는 예제를 통하여 보여주었듯이, 구조 예측 방법과 실험 방법이
함께 이용된다면 단백질 기능 해석에 관한 많 은 문제들을 효과적으로 해결 할 수 있을 것 이다.
참 고 문 헌
1. D. W. Mount, Bioinformatics: Sequence and Genome analysis, Cold Spring Habor Laboratory Press (2001).
2. D. L. Nelson and M. M. Cox, Lehninger Principles of Biochemistry (4th), W. H.
Freeman & Company (2004).
3. 연세대학교 구조생화학 및 분자생물학 연 구실, http://spin.yonsei.ac.kr/htsdnmr/.
4. 서울대학교 단백질 공학연구실, http://nmr.
snu.ac.kr.
5. M. J. E. Sternberg (editor), Protein Struc- ture Prediction: A Practical Approach, Oxford University Press (1997).
6. 김승연, 화학공학연구정보센터, 전문연구정 보, 단백질 구조 예측, http://www.
cheric.org/.
7. A. Fiser, R. K. Do, and A. Sali, Protein Science, 9, 1753 (2000).
8. M. A. Marti-Renom, A. Stuart, A. Fiser, R. Sanchez, F. Melo, and Sali, Annu.
Rev. Biophys. Biomol. Struct, 29, 291 (2000).
9. M. Tress, I. Ezkurdia, O. Garna, G. Lopez, and A. Valencia, Proteins: Structure, Function, and Bioinformatics Suppl., 7, 27 (2005).
10. G. Wang, Y. Jin, and R. L. Dunbrack Jr., Proteins: Structure, Function, and Bioinformatics Suppl., 7, 46 (2005).
11. J. J. Vincent, C. Tai, B. K. Sathyanarayana, and B. Lee, Proteins: Structure, Function, and Bioinformatics Suppl., 7, 67 (2005).
12. A. Kryshtafovych, C. Venclovas, K. Fidelis, and J. Moult, Proteins: Structure, Func- tion, and Bioinformatics Suppl., 7, 225 (2005).
13. Wikipedia, http://en.wikipedia.org/wiki/ CASP.
14. Y. S. Seo, A. Yoo, J. Jung, S. Sung, D.
% 저 자 소 개
유 아 림
2000 고려대학교 화학공학과 (학사) 2002 고려대학교 화학공학과 (석사) 2002∼현재 고려대학교 화공생명공학과
박사과정
이 원 태
1982 서울대학교 사범대학 물리학과 (학사)
1984 서울대학교 물리교육학과 (석사) 1992 University of Alabama at B'ham,
Dep. of Physics and Biochemistry (박사) 1992~1994 University of Toronto, Dep. of
Structural Biology (Postdoc) 1997~현재 연세대학교 생화학과 교수
R. Yang, W. T. Kim, and W. Lee, Biochem. J., 380, 339 (2004).
15. A. Yoo, Y. S. Seo, J. Jung, S. Sung, W.
T. Kim, D. R. Yang, and W. Lee, Journal of Structural Biology (in Press).
양 대 륙
1981 서울대학교 화학공학과 (학사) 1983 한국과학기술원 화학공학 (석사) 1990 University of California 화학공학
(박사)
1992~1994 포항공과대학교 화학공학과 조교수
1994~현재 고려대학교 화공생명공학과 교수