접수: 2012년 9월 26일, 승인: 2013년 1월 18일 연락처: 박병주, 110-750, 서울시 종로구 창경궁로 136
보령빌딩 5층
한국의약품안전관리원
Tel: (02) 2172-6710, Fax: (02) 2172-6701 E-mail: [email protected]
Correspondence to: Byung-Joo Park, MD, MPH, PhD Korea Institute of Drug Safety and Risk Management, 136 Changgyeonggung-ro, Jongno-gu, Seoul 110-750, Korea Tel: +82-2-2172-6710, Fax: +82-2-2172-6701
E-mail: [email protected]
미국의 약물안전성감시체계: 메드와치(MedWatch) 시스템을 중심으로
한국의약품안전관리원1, 서울대학교 의과대학 예방의학교실2
신선미1ㆍ정수연1ㆍ박병주1,2
An Overview of MedWatch System
Sun-Mi Shin, MSc
1, Sooyoun Chung, MD, PhD
1and Byung-Joo Park, MD, MPH, PhD
1,21
Korea Institute of Drug Safety and Risk Management,
2Department of Preventive Medicine, College of Medicine, Seoul National University, Seoul, Korea
Safety monitoring of medicinal products after marketing authorization is becoming a top priority for regulatory agencies and pharmaceutical industry. In the United States, initial post-marketing safety surveillance program began in the 1950s. MedWatch, Food and Drug Administration’s adverse drug reaction (ADR) reporting system, was founded in 1993. Information about voluntary reports by healthcare professionals and consumers, together with mandatory reports from manufacturers, has been accumulated in the Adverse Event Reporting System (AERS) from 1969. By the end of 2010, over 4 million reports are contained in this database. FDA systematically analyzes ADR reports, detects potential safety concerns, and manages them to protect the public health and minimize the risk of adverse drug reactions. Based on comprehensive evaluation of safety issues, the FDA has taken regulatory actions such as issuing safety alerts, restricting the use of the drug, ordering labeling changes, and communicating new information to the public. With advances in pharmacovigilance system accomplished for several decades, FDA is conducting anticipative management of drug safety issues. This paper will describe the architecture of the MedWatch system and explain the flow of data analysis and risk management. Finally it will suggest an outlook over future development of ADR reporting system in Korea. (JPERM 2013;6:73-78)
Key Words: MedWatch; Adverse drug reaction reports; Drug safety
서 론
의학의 발달과 함께 질병치료에 대한 기대감이 높아지면 서 약물안전성에 대한 관심은 크게 증가하고 있다. 약물감 시는 약물의 유해작용 또는 약물관련 문제의 탐지ㆍ평가ㆍ 해석ㆍ예방에 관한 과학적 연구와 활동을 말한다. 시판 전 임상시험은 한정된 수의 피험자를 대상으로 삼으면서 엄격 한 대상자의 선정기준을 적용하여 연령, 기저질환, 병용약
등이 매우 비현실적으로 단순화되어 있고, 투여기간과 관찰
기간이 짧다. 또한, 시험평가지표에만 관찰 초점을 두고 있
어 시판 후 불특정 다수가 다양한 환경에서 사용할 때에 대
한 현실적인 안전성정보를 얻기 어렵다.
1)따라서 시판 후
약물감시는 실제 상황에서 예상하지 못한 유해사례, 약물상
호작용, 의존도, 장기간의 효과, 약물내성 등 다양한 약물의
안전성관리를 할 수 있다는 점에서 그 중요성이 강조되어
왔다. 자발적 부작용보고는 시판 후 약물감시의 밑바탕으로
이를 위해 범국가적인 보고체계를 구축하여 활성화해오고 있다.
우리나라의 자발적 부작용보고제도는 1988년에 도입되었 고, 2006년부터 지역약물감시센터를 운영하고 2009년 약물 감시연구사업단을 출범하면서 그 보고건수가 급격히 증가 하였다. 2011년에는 66,395건, 2012년 상반기에는 37,544건 이 보고되어 2012년 6월 현재 누적건수가 202,974건으로 선 진국 수준으로 진입하고 있으나, 그동안 우리나라의 의약품 안전조치는 미국이나 유럽에 전적으로 의존하여 선제적 안 전성 조치는 취하지 못하고 있는 상황이다. 반면, 미국은 일 찍이 시판후 약물감시에 대한 중요성을 인지하고 1960년대 부터 국가적인 자발적 부작용보고제도를 실시하였다. 미국 식품의약품국은 제약회사가 제공하는 정보에 의존하는 것 이 아니라 유해사례보고자료를 활용해 안전성정보를 생산 하여 적극적으로 국민건강을 보호하기 위해 노력해왔다.
이에 미국의 약물감시체계 중 자발적 부작용보고를 바탕 으로 한 안전성 정보관리체계 및 위해관리 현황을 중점적으 로 살펴봄으로써 국내 약물안전성감시프로그램의 발전방향 을 모색하고자 한다.
1. 미국의 약물감시 조직체계
미국식품의약품국 조직은 의약품의 안전하고 효과적인 사용을 위해 분야별로 전문화되고 상호협력적인 체계로 구 성되어 있다. 미국식품의약품국은 6개의 센터로 구성되어 있으며, 주로 의약품(처방의약품, 일반의약품 포함)의 유효 성과 안전성을 관리, 감독하는 업무는 의약품평가연구센터 (center for drug evaluation and research, CDER)에서 담당하 며, 혈액제제 및 백신과 같은 생물학적제제에 대해서는 생물 학적제제평가연구센터(center for biologics evaluation and re- search, CBER), 의료기기는 의료기기ㆍ방사선보건센터(center for devices and radiological health, CDRH)에서 담당한다. 의약 품평가연구센터는 다시 12개의 부서로 구성되어 있는데, 이 중 의약품의 안전성평가에 깊이 관여하는 부서는 신약심사과 (office of new drugs, OND)와 위해감시ㆍ약물역학과(office of surveillance and epidemiology, OSE)를 꼽을 수 있다. 신약심 사과는 개발중인 임상시험용의약품을 관리하고 시판 전 단계 에서 의약품의 유효성과 안전성을 심사하여 시판 허가 여부 를 결정하는 업무를 하는 반면, 위해감시ㆍ약물역학과는 시판 중인 의약품의 안전성을 관리하며 유해사례를 줄이고 올바른 의약품 사용을 위한 위해관리 업무를 한다. 위해감시ㆍ약물역 학과는 본래 약물역학ㆍ통계과(office of pharmacoepidemiolo- gy and statistical science, OPaSS)에 소속된 약물안전과(office of drug safety, ODS)였으나, 시판 후 약물감시의 중요성이 점점 강조되면서 2006년 신약심사과와 동격인 상위 부서로 격상되었고, 그 명칭이 현 위해감시ㆍ약물역학과로 변경되
었다. 더불어, 2007년 123명에서 2012년 4월 현재 245명으로 그 규모가 2배로 확장되었으며, 의약품의 시판 전부터 시판 후까지 총체적인 안전성관리를 위해 신약심사과와 동등한 입장에서 조직적인 협력체계를 이루도록 하고 있다.
2)의약 품평가연구센터 내부규정(manual of policies and procedures, MAPP)에는 2개 과가 안전성문제에 대한 정보를 서로 공유 하고 조치 방안에 대한 의견을 수렴하기 위해 정기적으로(2 달에 1회 이상) 안전성합동회의(joint safety meeting)를 갖도 록 하고 있으며, 회의 참여자 및 이들의 역할 분담, 주요 안 건, 회의의 상세과정 및 일정 등을 명시하고 있다.
3)또한, 안 전성문제 발생시 상시적으로 안전성이슈팀(safety issue team) 을 만들어 각 부서간의 의견을 조율하고 최선의 해결 방법 을 찾도록 하고 있으며, 2009년 양 부서간 양해각서(memo- randum of agreement)를 교환하여 의약품 안전성관리에 대해 동등한 책임과 의무를 가지게 되었다.
위해감시ㆍ약물역학과는 세부적으로 5개의 계가 있는데, 잠재적인 안전성정보의 검색과 평가를 하는 약물감시연구1 계, 약물감시연구2계, 역학연구를 검토하는 역학계, 투약오 류분석ㆍ예방계, 약물위해관리계로 구성되어있다. 안전성평 가업무는 주로 의사, 약사, 간호사들로 구성된 안전성평가 관과 의무사무관이 담당하며, 역학자, 보건학자, 위해관리분 석가, 사회학자, 정보기술자, 규제전문가 등 다양한 분야의 전문가들이 있어 안전성문제 도출시 각 분야별 전문가들로 팀을 구성해 최선의 조치방법을 찾는다.
이러한 조직체계와 협력구조는 시판 후 약물감시가 한 부 서의 단독업무로 이루어지는 것이 아니라 개발단계 전부터 유기적으로 연결되며, 의학, 약학, 통계학, 사회학 등 다양한 관점에서 종합적인 의견이 수렴되어야 하는 복합적인 분야 임을 강조하고 있다.
2. 의약품 유해사례보고자료의 수집, 검토
미국이 의약품에 대하여 시판 후 안전성모니터링에 대해 관심을 가진 것은 1949년 초반 광범위 항생제인 클로람페니 콜의 재생불량성 빈혈과의 관련성이 보고되면서 시작되었 다. 1954년 미국의사협회와 함께 처음으로 의약품 시판후감 시프로그램(postmarketing surveillance program)을 설립하여 이상혈액질환에 대한 파일럿프로그램을 실시하였으며, 1961 년 모든 의약품의 유해사례를 모니터링하는 것으로 확대하 였다. 이후 1961년 탈리도마이드 사건을 계기로 자체 약물 유해반응모니터링사업을 결정하면서 미국의사협회의 프로 그램은 미국식품의약품국으로 이관되었다.
4)1962년, Kefauver Harris 개정법이 제정된 후부터 제약회사
는 신약허가신청을 거친 모든 제품에 대하여 시판 후 유해
사례를 보고하는 의무를 가지게 되었으며, 1964년부터는 보
건의료인도 약물의 안전성정보를 보고하도록 권장하게 되
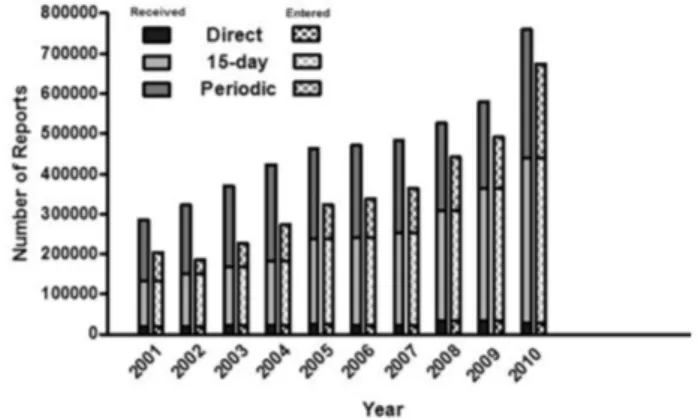
Figure 1. The number of reports received (solid bars) and entered
(checked bars) into AERS by type of report since the year 2000 until the end of 2010. Reprinted from Reports Received and Re- ports Entered into AERS by Year, Retrieved July 20, 2012, from http://www.fda.gov/Drugs/Guidance ComplianceRegulatoryInformat- ion/Surveillance/AdverseDrugEffects/ucm070434.htm.었다. 자발적유해사례보고를 활성화하기 위해 기존 5가지 유해사례보고양식을 ‘FDA 양식 3500’으로 통일하며 1993년 부터 메드와치 프로그램을 시작하였다. 메드와치는 의약품 유해사례를 수집하기 위한 수단이면서 안전성정보를 보건 의료인과 소비자에게 알리는 채널로, 약물감시를 위해 규제 기관과 대중이 만나는 소통공간이라고 말할 수 있다. 소비 자와 보건의료인은 자발적으로 유해사례보고양식을 작성하 여 제출하거나 1998년부터는 온라인보고시스템을 통해 직 접 보고할 수 있다.
반면, 제약회사는 의무적으로 유해사례를 보고하도록 규 정하고 있다. 제약회사는 매년 정기적으로 미국식품의약품 국의 각 해당부서에 보고해야 하는데, 신물질의 경우 승인 후 3년동안은 분기별로 보고하도록 하여 신약에 대해 집중 적인 모니터링을 하고 있다. 또한, 1998년부터는 신속보고 제도를 도입하여 예상치 못한(허가사항에 없는) 중대한 유 해사례에 대해서는 알게 된 후 15일 이내 의무보고 하도록 규정하고 있다. 현재 제약회사의 정기보고 및 신속보고는 전체 유해사례보고의 대부분인 약 95% 이상을 차지한다.
미국식품의약품국의 유해사례 수집 및 검토 목적은 주로 중대한 유해사례의 관리에 초점을 맞추고 있다. 메드와치는 중대한 유해사례의 의미를 명확하게 소개하고 이에 대한 보 고를 많이 할 수 있도록 권장하고 있다. 특히 소비자에게 중 대한 사례가 발생했을 때 보건의료인과의 상담을 통해 적절 한 의학적 정보를 얻고 이를 바탕으로 보고양식을 작성하길 권장함으로써 보고의 질을 높이기 위해 노력하고 있다.
5)제 약회사의 경우 예상하지 못한 중대한 유해사례에 대해서는 15일이내 보고해야 하는 의무를 부여하는 한편, 예상할 수 있고(허가사항에 기재되어 있고) 중대하지 않은 유해사례에 대해서는 보고를 면제받을 수 있는 기회를 준다. 단, 이 경 우 제약회사 내부적으로는 지속적인 유해사례 관리가 이루 어져야 하며, 미국식품의약품국 요구 시 5일 이내 그 자료 를 제출하도록 명시하고 있다.
6)유해사례가 보고되면 개인정보를 삭제하고 내용의 충실 성과 코딩의 정확성을 검토한 후 데이터베이스에 입력되는 데, 이 과정에서도 중대한 유해사례는 집중관리대상이 된 다. 보고자료 중 직접보고와 15일 신속보고는 우선적인 검 토대상이 되어 위해감시ㆍ약물역학과의 안전성평가 담당자 들이 먼저 검토하고 표준화작업을 거쳐 14일 이내 데이터베 이스에 입력한다. 반면 정기보고는 분기별로 검토된 후 데 이터베이스에 입력된다.
미국식품의약품국 자료에 따르면, 2010년 한 해 동안 데 이터베이스에 입력된 유해사례 총 673,259건 중 15일 신속 보고는 409,608건(60.8%)이며, 심각한 결과를 초래한 사례는 471,291건(70.0%)으로 전체 사례 중 중대한 경우가 많은 부 분을 차지하고 있다(그림 1).
7)중대한 유해사례에 대한 선택
과 집중 전략은, 상대적으로 경미하고 회복이 빠른 사례보 다 중대한 사례부터 우선적으로 찾아내어 최대한 피해를 줄 이고, 방대한 데이터베이스 속에서 그 발생빈도는 낮지만 심각한 수준의 경우를 신속하게 감지해내는 방법이 될 수 있다. 유해사례보고자료만으로는 사소한 안전성정보까지 모두 탐색하고 인과관계를 찾는 데는 한계가 존재하므로, 유해사례보고자료의 특성을 최대한 살려 이를 실용적으로 활용할 수 있는 방법에 집중하고 대중을 심각한 유해사례로 부터 우선적으로 보호한다는 측면에서 그 의미가 크다.
3. 유해사례보고자료의 활용 및 공개
미국은 1967년 유해사례보고자료 전산화를 도입한 후, 1968년
자발적보고시스템(spontaneous reporting system, SRS)을 구축하여
모든 유해사례를 코딩, 입력하였다. 1997년 11월에는 기존 자발적
보고시스템을 유해사례보고시스템(adverse event reporting system,
AERS)으로 대체하여 1969년부터 현재까지의 허가된 모든
의약품과 생물학적제제 등에 대한 유해사례데이터베이스를
보유하고 있다. 통계자료에 의하면 2005년에는 323,384건,
2010년에는 673,259건이 보고되어 최근 5년 사이 유해사례 보
고량은 2배 이상 증가하였다. 2010년까지 유해사례보고시스
템에는 약 400만건 이상의 데이터가 누적되어 있으며, 2011년
약 80만여건으로 추정되는 보고건을 더하면 매년 급속한 팽
창속도를 짐작할 수 있다. 의약품평가연구센터와 생물학적
제제평가연구센터에서는 방대한 유해사례보고자료를 바탕
으로 데이터마이닝기법을 적용하여 약물과의 인과관계 가능
성이 있다고 검색되는 실마리정보를 평가한다. 2007년 미국
식품의약품국개정법령(FDA amendment act of 2007, FDAAA)
에 근거하여 2008년부터는 2주에 한번 주기적으로 실마리
정보를 검색하고, 잠재적 실마리정보와 새로운 안전성정보
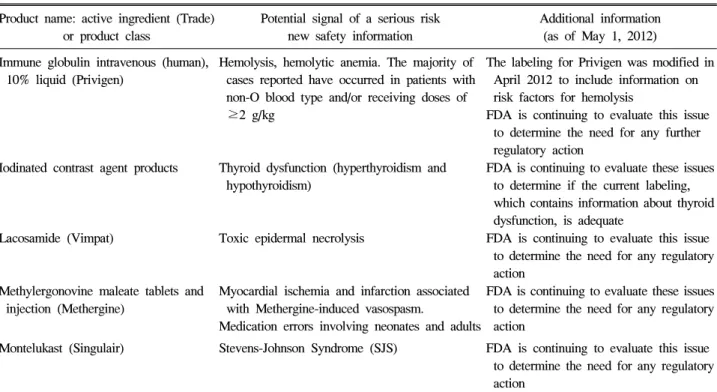
Table 1. An example of potential signals of serious risks/new safety information identified by the Adverse Event Reporting System (AERS)
Product name: active ingredient (Trade) or product class
Potential signal of a serious risk new safety information
Additional information (as of May 1, 2012) Immune globulin intravenous (human),
10% liquid (Privigen)
Hemolysis, hemolytic anemia. The majority of cases reported have occurred in patients with non-O blood type and/or receiving doses of
≥2 g/kg
The labeling for Privigen was modified in April 2012 to include information on risk factors for hemolysis
FDA is continuing to evaluate this issue to determine the need for any further regulatory action
Iodinated contrast agent products Thyroid dysfunction (hyperthyroidism and hypothyroidism)
FDA is continuing to evaluate these issues to determine if the current labeling, which contains information about thyroid dysfunction, is adequate
Lacosamide (Vimpat) Toxic epidermal necrolysis FDA is continuing to evaluate this issue to determine the need for any regulatory action
Methylergonovine maleate tablets and injection (Methergine)
Myocardial ischemia and infarction associated with Methergine-induced vasospasm.
Medication errors involving neonates and adults
FDA is continuing to evaluate these issues to determine the need for any regulatory action
Montelukast (Singulair) Stevens-Johnson Syndrome (SJS) FDA is continuing to evaluate this issue to determine the need for any regulatory action
Reprinted from Potential Signals of Serious Risks/New Safety Information identified by the AERS between January-March 2012, Retrieved July 20, 2012, from http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Surveillance/AdverseDrugEffects/ucm307608.htm.
에 대해 분기별로 웹페이지에 공개하고 있다(Title IX, Section 921, FDAAA). 이 공개자료에는 약물정보, 잠재적 실 마리정보 또는 새로운 안전성정보, 조치내용(허가사항 변경, 허가 취소, 평가 중인 내용 등)이 담겨있으며, 추후 새로운 안전성정보가 발견되어 조치를 바꿀 경우 그 내용을 지속적 으로 수정한다. 평가 및 조치가 완료된 실마리정보 뿐 아니라 평가 중인 내용을 공개하는 것은, 유해사례를 보고한 대중에 게 도출된 정보를 투명하게 공개하고 생산된 결과를 환원하 는 의미이기도 하며, 유의미한 안전성정보가 아닐 지라도 환 자 또는 의료인에게 사전에 주의할 수 있는 기회를 주어 위 해를 줄이고자 하는 의지로 해석된다. 단, 불필요한 오해를 줄이기 위해, 이 자료는 처방금지 또는 복용금지를 의미하는 것이 아니라 잠재적인 안전성정보일 뿐임을 강조하여 함께 명시하고 있다(표 1).
8)데이터베이스에서 심각한 유해사례가 실마리정보로 검색되었을 경우 의약품평가연구센터의 문서 보관ㆍ보고ㆍ규제추적시스템(document archiving, reporting, and regulatory tracking system, DARRTS) 또는 생물학적제제평가연구 센터의 치료ㆍ혈액제제안전성추적시스템(therapeutics and blood safety branch safety signal tracking system)에 안전성이슈(trac- ked safety issus, TSI)로 입력하여 지속적으로 관리하며, 이를 외부 유관 부서들과도 공유하여 평가 및 조치사항을 결정한 다.
9)또한, 분기마다 실마리정보의 내용을 결정하고 사전에
의견을 수렴하기 위해 각 부서의 담당자들로 구성된 921팀 을 별도로 구성하고 규정된 절차에 따라 진행하도록 함으로 써 실마리정보의 평가 및 공개에 신중을 기하고 있다.
10)미국식품의약품국은 실마리정보를 공개할 뿐 아니라 유 해사례보고자료 자체를 두가지 형태로 공개한다. 첫 번째 형태는, 유해사례보고시스템 데이터베이스로부터 분기별로 유해사례를 추출하여 공개하는 자료인데, ASCII나 SGML 형태의 파일로 압축되어 있어 개개의 보고자료 원문을 그대 로 볼 수는 없지만, 기본정보, 약물정보, 유해사례정보, 유해 사례결과 등의 데이터 항목을 담고 있어 통계프로그램을 통 해 누구나 이 자료의 이용이 가능하다.
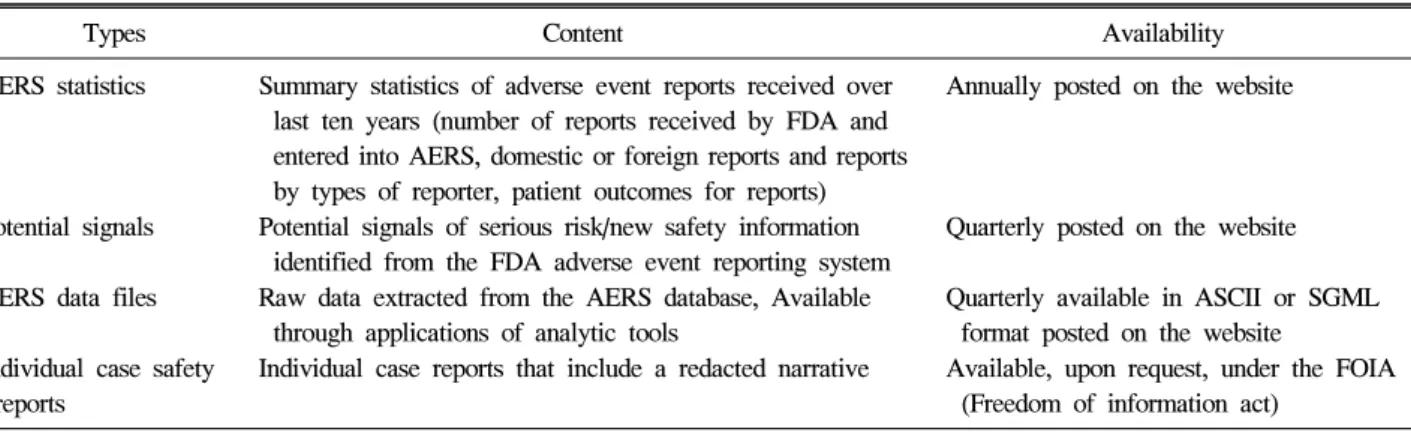
11)두 번째 형태는 개 개의 보고자료 형태로서, 정보자유법 요청양식(freedom of information act request form)에 원하는 정보사항을 기재하여 제출하면 개인정보가 삭제된 원자료를 추출하여 제공해준 다. 개인정보를 보호하는 범위 안에서 유해사례보고자료를 공개하는 것은 국민의 알권리를 보장할 뿐만 아니라, 정책 결정에 투명성을 부여하여 신뢰도를 높일 수 있다는 데에 그 의의가 크다고 할 수 있다(표 2).
4. 미국식품의약품국의 시판 후 약물감시 성과 및 방향
2000년대로 접어들면서, 의약품 안전관리를 강화하고 안
전성평가방법에 최신의 기술을 도입하고자 하는 변화의 바
Table 2. Types of adverse event reporting system (AERS) data available to the public
Types Content Availability
AERS statistics Summary statistics of adverse event reports received over last ten years (number of reports received by FDA and entered into AERS, domestic or foreign reports and reports by types of reporter, patient outcomes for reports)
Annually posted on the website
Potential signals Potential signals of serious risk/new safety information identified from the FDA adverse event reporting system
Quarterly posted on the website
AERS data files Raw data extracted from the AERS database, Available through applications of analytic tools
Quarterly available in ASCII or SGML format posted on the website
Individual case safety reports
Individual case reports that include a redacted narrative Available, upon request, under the FOIA (Freedom of information act)
람이 불기 시작했다. 2004년 11월, 미국식품의약품국은 의 약품 안전관리강화계획을 발표하였고, 안전관리프로그램에 대한 점검을 위해 의학한림원(institute of medicine, IOM)에 평가를 요청하였다. 이에 2006년 의학한림원이 제출한 보고 서에는 시판 후 안전성관리의 취약점과 보강의 필요성을 피 력하고, 이를 위한 미국식품의약품국의 법률적 권한 강화를 강조하는 내용이 담겨있었으며, 2007년 미국식품의약품국 개정법령(FDA amendment act, FDAAA)에 상당부분 반영되 어 시행되었다.
이전에는 허가사항 변경이나 시판 후 임상시험이 주로 제약회사의 자발적인 실시로 이루어졌지만, 이러한 제도변 화를 기반으로 제약회사에 안전성관리를 요구할 수 있는 권 한을 가지게 되었다. 그 결과, 2008년 이후 제약회사에 약 385회 시판 후 안전성평가연구의 실시를 요구하였고, 새로 운 안전성정보를 허가사항에 반영하도록 약물군 전체를 대 상으로 약 65회의 변경지시를 내렸다. 또한, 기존 약물과 신 약 대상으로 64개의 위해평가ㆍ완화전략(risk evaluation and mitigation strategy, REMS) 프로그램 실시를 요구해왔다.
2)이 러한 행정적 조치 중 상당 부분은 유해사례보고자료에서 도 출된 실마리정보를 바탕으로 이루어졌으며,
12)지속적으로 선제적인 안전성관리를 위해 유해사례보고의 양적ㆍ질적 향상 및 실마리정보 평가방법의 개선을 위해 노력하고 있 다. 현재 진행중인 유해사례보고 전산시스템의 업그레이드 와 재현성있고 객관적인 안전성평가가이드라인 개발은 그 일환이라고 할 수 있다.
13)더불어, 유해사례보고자료 및 기 구축된 대규모 전산자료 연계를 통해 능동적인 약물감시체 계를 구현하고자 하고 있다.
긴 역사를 가지고 있는 메드와치에 비해 우리나라의 유 해사례보고시스템은 아직 초기단계이다. 지역약물감시센터 와 약물감시연구사업단의 출범으로 보고건수가 단기간에 급증하며 놀라운 성장 속도를 보이고 있으나, 소비자 또는 보건의료인의 자발적인 보고문화는 아직 정착하지 못하고
있는 실정이다. 이러한 시점에서 설립된 한국의약품안전관 리원은 의약품의 안전성정보를 수집ㆍ분석ㆍ관리하는데 있 어서 의약품유해사례보고시스템을 선진국 수준으로 구축하 는 것이 매우 중요하다고 생각한다. 유해사례보고의 중요성 및 보고방법을 교육하고 홍보하는 활동이 확대되어야 하며, 산출된 안전성정보를 대중에게 공개하고 피드백을 줌으로 써 보고의 지속적인 동기 부여가 되어, 대중과 소통하는 선 순환적인 약물감시체계로의 발전이 필요하다. 더불어, 유해 사례보고자료를 활용한 약물감시프로그램을 한국의약품안 전관리원이 우리나라 실정에 맞게 단계적으로 확립하고, 여 기에 제도적, 기술적 인프라를 접목하여 선진적인 의약품안 전관리체계로 발전시켜 나가길 기대한다.
참고문헌
1. Strom BL. Pharmacoepidemiology, 5th ed. Chichester: John Wiley & Sons, Ltd.; 2012. p. 13-6.
2. Advances in FDA’s Safety Program for Marketed Drugs.
[cited 2012 April]; Available from: http://www.fda.gov/Drugs/
DrugSafety/ucm297389.htm [accessed 2012 July 20].
3. Joint safety meetings between OND and OSE (CDER MAPP 4151.7). Available from: http://www.fda.gov/AboutFDA/Cen- tersOffices/OfficeofMedicalProductsandTobacco/CDER/Manu- alofPoliciesProcedures/default.htm [accessed 2012 July 20].
4. Ropp KL. MedWatch: On The Lookout For Medical Product Problems. FDA Consumer 1993;27:14-7.
5. Reporting by consumers. Available from: http://www.fda.gov/
Safety/MedWatch/HowToReport/ucm053074.htm [accessed 2012 July 20].
6. Postmarketing adverse experience reporting for human drug and licensed biological products: FDA guidance for industry. Available from: http://www.fda.gov/Drugs/DrugSafety/ucm299833.htm [accessed 2012 July 20].
7. Adverse event reporting system (AERS) statistics. Available from:
http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformati on/Surveillance/AdverseDrugEffects/ucm070093.htm [accessed 2012 July 20].
8. Potential Signals of Serious Risks/New Safety Information Identified by the Adverse Event Reporting System (AERS).
Available from: http://www.fda.gov/Drugs/Guidance Compli- anceRegulatoryInformation/Surveillance/AdverseDrug Effects/ucm307608.htm [acceessed 2012 July 20].
9. Tracking of significant safety issues in marketed drugs? Use of the DARRTS Tracked safety issues (CDER MAPP 4121.2).
Available from: http://www.fda.gov/AboutFDA/CentersOffices/
OfficeofMedicalProductsandTobacco/CDER/ManualofPoliciesPr ocedures/default.htm [accessed 2012 July 20].
10. FDA Posting of Potential Signals of Serious Risks Identified by
the Adverse Event Reporting System (CDER MAPP 6700.9).
Available from: http://www.fda.gov/AboutFDA/CentersOffi- ces/OfficeofMedicalProductsandTobacco/CDER/Manualof- PoliciesProcedures/default.htm [accessed 2012 July 20].
11. The Adverse Event Reporting System (AERS): Latest Quar- terly Data Files. Available from: http://www.fda.gov/Drugs/
GuidanceComplianceRegulatoryInformation/Surveillance/Ad- verseDrugEffects/ucm082193.htm [accessed 2012 July 20]
12. Powers A, Cook GE. Potential Safety Signals and Their Sig- nificance. Arch Intern Med 2012;172:72-3.
13. FDA science board subcommittee: Review of the FDA/CDER pharmacovigilance program 2011. Available from: http://www.
fda.gov/AdvisoryCommittees/CommitteesMeetingMaterials/
ScienceBoardtotheFoodandDrugAdministration/ucm241888.htm [accessed 2012 July 20].